What is the cause of rett syndrome
Rett Syndrome Fact Sheet | National Institute of Neurological Disorders and Stroke
What is Rett syndrome?
What are the stages of the disorder?
What causes Rett syndrome?
Is Rett syndrome inherited?
Who gets Rett syndrome?
How is Rett syndrome diagnosed?
Is treatment available?
What is the outlook for those with Rett syndrome?
What research is being done?
Where can I get more information?
What is Rett syndrome?
Rett syndrome is a neurodevelopmental disorder that affects girls almost exclusively. It is characterized by normal early growth and development followed by a slowing of development, loss of purposeful use of the hands, distinctive hand movements, slowed brain and head growth, problems with walking, seizures, and intellectual disability.
The disorder was identified by Dr. Andreas Rett, an Austrian physician who first described it in a journal article in 1966. It was not until after a second article about the disorder, published in 1983 by Swedish researcher Dr. Bengt Hagberg, that the disorder was generally recognized.
The course of Rett syndrome, including the age of onset and the severity of symptoms, varies from child to child. Before the symptoms begin, however, the child generally appears to grow and develop normally, although there are often subtle abnormalities even in early infancy, such as loss of muscle tone (hypotonia), difficulty feeding, and jerkiness in limb movements. Then, gradually, mental and physical symptoms appear. As the syndrome progresses, the child loses purposeful use of her hands and the ability to speak. Other early symptoms may include problems crawling or walking and diminished eye contact. The loss of functional use of the hands is followed by compulsive hand movements such as wringing and washing. The onset of this period of regression is sometimes sudden.
Apraxia — the inability to perform motor functions — is perhaps the most severely disabling feature of Rett syndrome, interfering with every body movement, including eye gaze and speech.
Children with Rett syndrome often exhibit autistic-like behaviors in the early stages. Other symptoms may include walking on the toes, sleep problems, a wide-based gait, teeth grinding and difficulty chewing, slowed growth, seizures, cognitive disabilities, and breathing difficulties while awake such as hyperventilation, apnea (breath holding), and air swallowing.
top
What are the stages of the disorder?
Scientists generally describe four stages of Rett syndrome. Stage I, called early onset, typically begins between 6 and 18 months of age. This stage is often overlooked because symptoms of the disorder may be somewhat vague, and parents and doctors may not notice the subtle slowing of development at first. The infant may begin to show less eye contact and have reduced interest in toys. There may be delays in gross motor skills such as sitting or crawling. Hand-wringing and decreasing head growth may occur, but not enough to draw attention. This stage usually lasts for a few months but can continue for more than a year.
Stage II, or the rapid destructive stage, usually begins between ages 1 and 4 and may last for weeks or months. Its onset may be rapid or gradual as the child loses purposeful hand skills and spoken language. Characteristic hand movements such as wringing, washing, clapping, or tapping, as well as repeatedly moving the hands to the mouth often begin during this stage. The child may hold the hands clasped behind the back or held at the sides, with random touching, grasping, and releasing. The movements continue while the child is awake but disappear during sleep. Breathing irregularities such as episodes of apnea and hyperventilation may occur, although breathing usually improves during sleep. Some girls also display autistic-like symptoms such as loss of social interaction and communication. Walking may be unsteady and initiating motor movements can be difficult. Slowed head growth is usually noticed during this stage.
Stage III, or the plateau or pseudo-stationary stage, usually begins between ages 2 and 10 and can last for years. Apraxia, motor problems, and seizures are prominent during this stage. However, there may be improvement in behavior, with less irritability, crying, and autistic-like features. A girl in stage III may show more interest in her surroundings and her alertness, attention span, and communication skills may improve. Many girls remain in this stage for most of their lives.
Apraxia, motor problems, and seizures are prominent during this stage. However, there may be improvement in behavior, with less irritability, crying, and autistic-like features. A girl in stage III may show more interest in her surroundings and her alertness, attention span, and communication skills may improve. Many girls remain in this stage for most of their lives.
Stage IV, or the late motor deterioration stage, can last for years or decades. Prominent features include reduced mobility, curvature of the spine (scoliosis) and muscle weakness, rigidity, spasticity, and increased muscle tone with abnormal posturing of an arm, leg, or top part of the body. Girls who were previously able to walk may stop walking. Cognition, communication, or hand skills generally do not decline in stage IV. Repetitive hand movements may decrease and eye gaze usually improves.
top
What causes Rett syndrome?
Nearly all cases of Rett syndrome are caused by a mutation in the methyl CpG binding protein 2, or MECP2 (pronounced meck-pea-two) gene. Scientists identified the gene — which is believed to control the functions of many other genes — in 1999. The
MECP2 gene contains instructions for the synthesis of a protein called methyl cytosine binding protein 2 (MeCP2), which is needed for brain development and acts as one of the many biochemical switches that can either increase gene expression or tell other genes when to turn off and stop producing their own unique proteins. Because theMECP2 gene does not function properly in individuals with Rett syndrome, insufficient amounts or structurally abnormal forms of the protein are produced and can cause other genes to be abnormally expressed.
Scientists identified the gene — which is believed to control the functions of many other genes — in 1999. The
MECP2 gene contains instructions for the synthesis of a protein called methyl cytosine binding protein 2 (MeCP2), which is needed for brain development and acts as one of the many biochemical switches that can either increase gene expression or tell other genes when to turn off and stop producing their own unique proteins. Because theMECP2 gene does not function properly in individuals with Rett syndrome, insufficient amounts or structurally abnormal forms of the protein are produced and can cause other genes to be abnormally expressed.
Not everyone who has an MECP2 mutation has Rett syndrome. Scientists have identified mutations in the CDKL5 andFOXG1 genes in individuals who have atypical or congenital Rett syndrome, but they are still learning how those mutations cause the disorder. Scientists believe the remaining cases may be caused by partial gene deletions, mutations in other parts of the MECP2 gene, or additional genes that have not yet been identified, and they continue to look for other causes.
top
Is Rett syndrome inherited?
Although Rett syndrome is a genetic disorder, less than 1 percent of recorded cases are inherited or passed from one generation to the next. Most cases are spontaneous, which means the mutation occurs randomly. However, in some families of individuals affected by Rett syndrome, there are other female family members who have a mutation of theirMECP2 gene but do not show clinical symptoms. These females are known as “asymptomatic female carriers.”
top
Who gets Rett syndrome?
Rett syndrome is estimated to affect one in every 10,000 to 15,000 live female births and in all racial and ethnic groups worldwide. Prenatal testing is available for families with an affected daughter who has an identified MECP2 mutation. Since the disorder occurs spontaneously in most affected individuals, however, the risk of a family having a second child with the disorder is less than 1 percent.
Genetic testing is also available for sisters of girls with Rett syndrome who have an identified MECP2 mutation to determine if they are asymptomatic carriers of the disorder, which is an extremely rare possibility.
The MECP2 gene is found on a person’s X chromosome, one of the two sex chromosomes. Girls have two X chromosomes, but only one is active in any given cell. This means that in a girl with Rett syndrome only a portion of the cells in the nervous system will use the defective gene. Some of the child's brain cells use the healthy gene and express normal amounts of the protein.
The severity of Rett syndrome in girls is in part a function of the percentage of their cells that express a normal copy of the MECP2 gene. If the active X chromosome that is carrying the defective gene is turned off in a large proportion of cells, the symptoms will be mild, but if a larger percentage of cells have the X chromosome with the normal MECP2 gene turned off, onset of the disorder may occur earlier and the symptoms may be more severe.
The story is different for boys who have a MECP2 mutation known to cause Rett syndrome in girls. Because boys have only one X chromosome (and one Y chromosome) they lack a back-up copy that could compensate for the defective one, and they have no protection from the harmful effects of the disorder. Boys with such a defect frequently do not show clinical features of Rett syndrome but experience severe problems when they are first born and die shortly after birth. A very small number of boys may have a different mutation in the MECP2 gene or a sporadic mutation after conception that can cause some degree of intellectual disability and developmental problems.
Boys with such a defect frequently do not show clinical features of Rett syndrome but experience severe problems when they are first born and die shortly after birth. A very small number of boys may have a different mutation in the MECP2 gene or a sporadic mutation after conception that can cause some degree of intellectual disability and developmental problems.
top
How is Rett syndrome diagnosed?
Doctors clinically diagnose Rett syndrome by observing signs and symptoms during the child's early growth and development, and conducting ongoing evaluations of the child's physical and neurological status. Scientists have developed a genetic test to complement the clinical diagnosis, which involves searching for the MECP2 mutation on the child's X chromosome.
A pediatric neurologist, clinical geneticist, or developmental pediatrician should be consulted to confirm the clinical diagnosis of Rett syndrome. The physician will use a highly specific set of guidelines that are divided into three types of clinical criteria: main, supportive, and exclusion.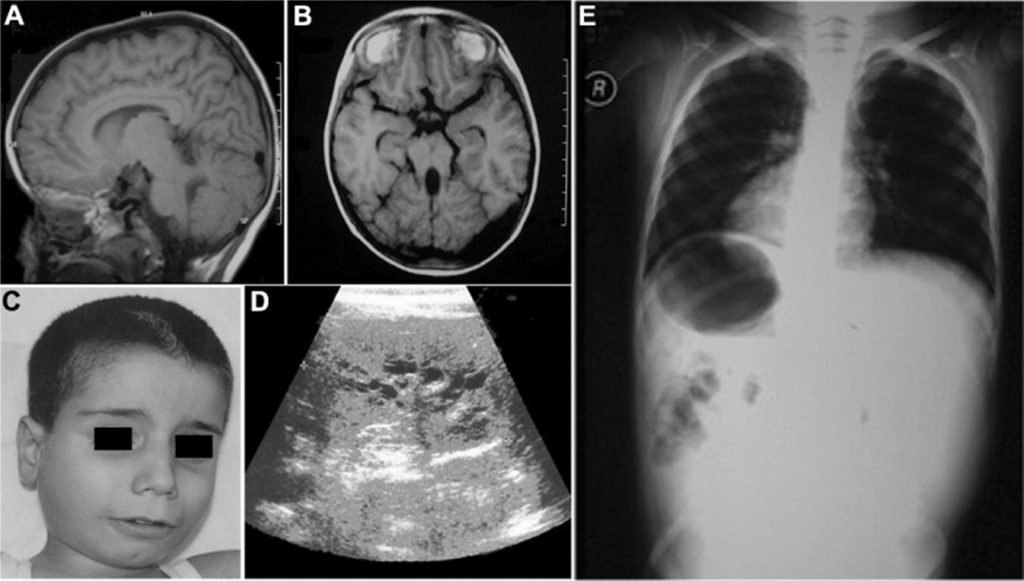 The presence of any of the exclusion criteria negates a diagnosis of classic Rett syndrome.
The presence of any of the exclusion criteria negates a diagnosis of classic Rett syndrome.
Examples of main diagnostic criteria or symptoms include partial or complete loss of acquired purposeful hand skills, partial or complete loss of acquired spoken language, repetitive hand movements (such has hand wringing or squeezing, clapping or rubbing), and gait abnormalities, including toe-walking or an unsteady, wide-based, stiff-legged walk.
Supportive criteria are not required for a diagnosis of Rett syndrome but may occur in some individuals. In addition, these symptoms — which vary in severity from child to child — may not be observed in very young girls but may develop with age. A child with supportive criteria but none of the essential criteria does not have Rett syndrome. Supportive criteria include scoliosis. teeth-grinding, small cold hands and feet in relation to height, abnormal sleep patterns, abnormal muscle tone, inappropriate laughing or screaming, intense eye communication, and diminished response to pain.
In addition to the main diagnostic criteria, a number of specific conditions enable physicians to rule out a diagnosis of Rett syndrome. These are referred to as exclusion criteria. Children with any one of the following criteria do not have Rett syndrome: brain injury secondary to trauma, neurometabolic disease, severe infection that causes neurological problems; and grossly abnormal psychomotor development in the first 6 months of life.
top
Is treatment available?
There is no cure for Rett syndrome. Treatment for the disorder is symptomatic — focusing on the management of symptoms — and supportive, requiring a multidisciplinary approach. Medication may be needed for breathing irregularities and motor difficulties, and anticonvulsant drugs may be used to control seizures. There should be regular monitoring for scoliosis and possible heart abnormalities. Occupational therapy can help children develop skills needed for performing self-directed activities (such as dressing, feeding, and practicing arts and crafts), while physical therapy and hydrotherapy may prolong mobility. Some children may require special equipment and aids such as braces to arrest scoliosis, splints to modify hand movements, and nutritional programs to help them maintain adequate weight. Special academic, social, vocational, and support services may be required in some cases.
Some children may require special equipment and aids such as braces to arrest scoliosis, splints to modify hand movements, and nutritional programs to help them maintain adequate weight. Special academic, social, vocational, and support services may be required in some cases.
top
What is the outlook for those with Rett syndrome?
Despite the difficulties with symptoms, many individuals with Rett syndrome continue to live well into middle age and beyond. Because the disorder is rare, very little is known about long-term prognosis and life expectancy. While there are women in their 40s and 50s with the disorder, currently it is not possible to make reliable estimates about life expectancy beyond age 40.
top
What research is being done?
Within the Federal government, the National Institute of Neurological Disorders and Stroke (NINDS), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Institute of Mental Health (NIMH), and the Office of Rare Diseases Research (ORDR) support clinical and basic research on Rett syndrome.
Understanding the cause of this disorder is necessary for developing new therapies to manage specific symptoms, as well as for providing better methods of diagnosis. The discovery of the main Rett syndrome gene (MECP2) in 1999 provides a basis for further genetic studies and enables the use of recently developed animal models such as transgenic mice which are deficient in MECP2. These mice have neurologic abnormalities that can be reversed by activating the MECP2 gene later in life.
One NINDS-supported study looks for mutations in the MECP2 gene of individuals with Rett syndrome to learn about MeCP2 protein function and dysfunction. Information from this study will increase understanding of the disorder and may lead to new therapies. Other research aims at identifying molecular pathways that are affected by the dysfunction, developing animal models of the disorder, and early-stage therapy development.
Some researchers suggest that the specific type of mutation in the MECP2 gene affects the severity of symptoms of Rett syndrome.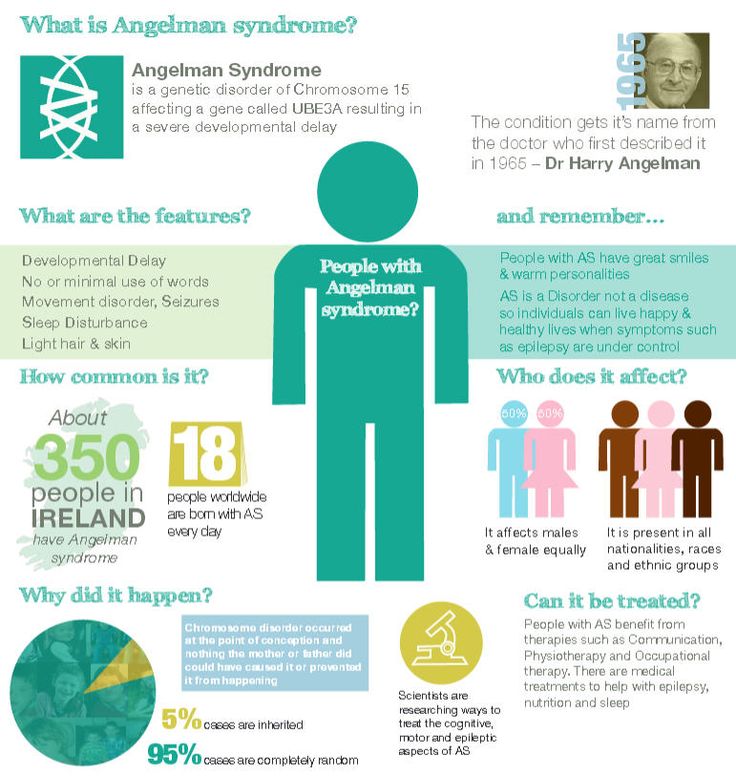 Studies are now underway to understand each mutation that may cause the features of Rett syndrome, and how these mutations might change the features of the syndrome. One NIH-funded study of the natural history of Rett syndrome should also provide new information about these topics.
Studies are now underway to understand each mutation that may cause the features of Rett syndrome, and how these mutations might change the features of the syndrome. One NIH-funded study of the natural history of Rett syndrome should also provide new information about these topics.
Scientists know that lack of a properly functioning MeCP2 protein disturbs the function of mature brain cells but they do not know the exact mechanisms by which this happens. Investigators are trying to find other genetic switches that operate in a similar way to the MeCP2 protein. Once they discover how the protein works and locate similar switches, they may devise therapies that can substitute for the malfunctioning switch. Another outcome might involve manipulating other biochemical pathways to compensate for the malfunctioning MECP2 gene, thereby preventing progression of the disorder. Gene therapy to achieve regulated expression of a normal MECP2 gene is also under study in animal models.
Researchers are also trying to find other genes that may be involved in Rett syndrome. Some studies have helped to narrow the search for these genes, but much is still unknown about how these genes may cause or contribute to Rett syndrome.
top
Where can I get more information?
For more information on neurological disorders or research programs funded by the National Institute of Neurological Disorders and Stroke, contact the Institute's Brain Resources and Information Network (BRAIN) at:
BRAIN
P.O. Box 5801
Bethesda, MD 20824
800-352-9424
Information also is available from the following organizations:
International Rett Syndrome Foundation
4600 Devitt Drive
Cincinnati, OH 45246
[email protected]
Tel: 513-874-1298; 800-818-7388
Easter Seals
233 South Wacker Drive
Suite 2400
Chicago, IL 60606
[email protected]
Tel: 312-726-6200; 800-221-6827
Fax: 312-726-1494
National Institute of Child Health and Human Development (NICHD)
National Institutes of Health, DHHS
31 Center Drive, Rm. 2A32 MSC 2425
2A32 MSC 2425
Bethesda, MD 20892-2425
Tel: 301-496-5133
Fax: 301-496-7101
National Institute of Mental Health (NIMH)
National Institutes of Health, DHHS
6001 Executive Blvd. Rm. 8184, MSC 9663
Bethesda, MD 20892-9663
[email protected]
Tel: 301-443-4513; 866-415-8051; 301-443-8431 (TTY)
Fax: 301-443-4279
Genetic and Rare Diseases Information Center (GARD)
National Institutes of Health, DHHS
P.O. Box 8126
Gaithersburg, MD 20898-8126
Tel: 888-205-2311
Rett Syndrome Research Trust
67 Under Cliff Road
Trumbull, CT 06611
[email protected]
Tel: 203-445-0041
"Rett Syndrome Fact Sheet", NINDS, Publication date November 2009.
NIH Publication No. 09-4863
Back to Rett Syndrome Information Page
See a list of all NINDS disorders
Publicaciones en Español
Síndrome de Rett
Prepared by:
Office of Communications and Public Liaison
National Institute of Neurological Disorders and Stroke
National Institutes of Health
Bethesda, MD 20892
NINDS health-related material is provided for information purposes only and does not necessarily represent endorsement by or an official position of the National Institute of Neurological Disorders and Stroke or any other Federal agency.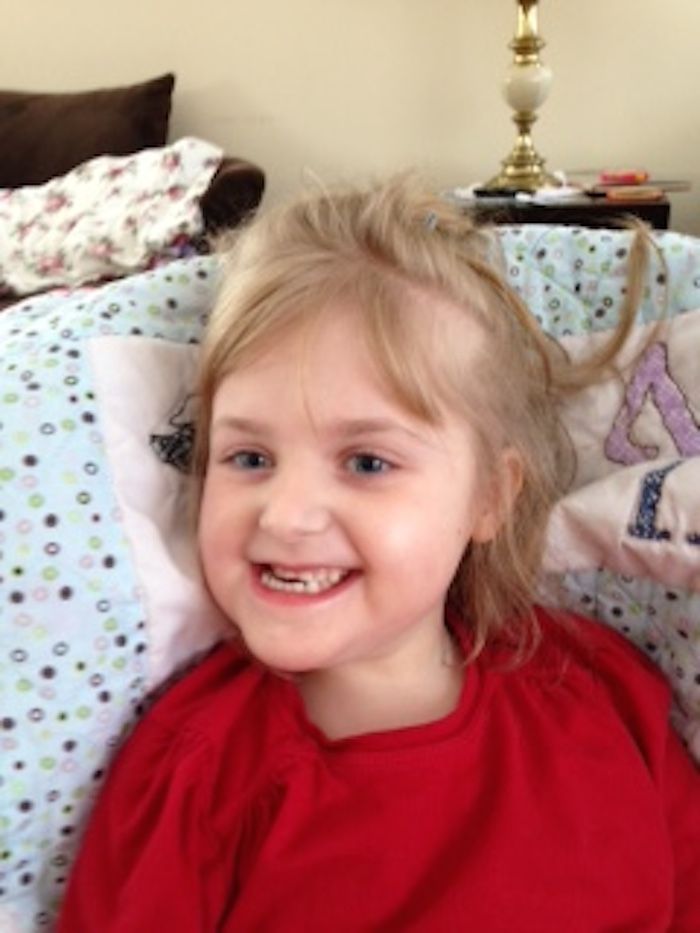 Advice on the treatment or care of an individual patient should be obtained through consultation with a physician who has examined that patient or is familiar with that patient's medical history.
Advice on the treatment or care of an individual patient should be obtained through consultation with a physician who has examined that patient or is familiar with that patient's medical history.
All NINDS-prepared information is in the public domain and may be freely copied. Credit to the NINDS or the NIH is appreciated.
Rett syndrome - Symptoms and causes
Overview
Rett syndrome is a rare genetic neurological and developmental disorder that affects the way the brain develops. This disorder causes a progressive loss of motor skills and language. Rett syndrome primarily affects females.
Most babies with Rett syndrome seem to develop as expected for the first six months of life. These babies then lose skills they previously had — such as the ability to crawl, walk, communicate or use their hands.
Over time, children with Rett syndrome have increasing problems with the use of muscles that control movement, coordination and communication. Rett syndrome can also cause seizures and intellectual disabilities. Unusual hand movements, such as repetitive rubbing or clapping, replace purposeful hand use.
Unusual hand movements, such as repetitive rubbing or clapping, replace purposeful hand use.
Although there's no cure for Rett syndrome, potential treatments are being studied. Current treatment focuses on improving movement and communication, treating seizures, and providing care and support for children and adults with Rett syndrome and their families.
Products & Services
- Book: Mayo Clinic Family Health Book, 5th Edition
- Newsletter: Mayo Clinic Health Letter — Digital Edition
Symptoms
Babies with Rett syndrome usually are born after an uncomplicated pregnancy and delivery. Most infants with Rett syndrome seem to grow and behave as expected for the first six months. After that, signs and symptoms start to appear.
The most pronounced changes generally occur at 12 to 18 months of age, over a period of weeks or months. Symptoms and their severity vary greatly from child to child.
The main signs and symptoms include:
- Slowed growth.
 Brain growth slows after birth. Smaller than usual head size (microcephaly) is sometimes the first sign that a child has Rett syndrome. As children get older, there is delayed growth in other parts of the body.
Brain growth slows after birth. Smaller than usual head size (microcephaly) is sometimes the first sign that a child has Rett syndrome. As children get older, there is delayed growth in other parts of the body. - Loss of movement and coordination abilities. The first signs often include reduced hand control and a decreasing ability to crawl or walk. At first, this loss of abilities occurs rapidly, and then it continues more gradually. Eventually muscles become weak or stiff, with unusual movement and positioning.
- Loss of communication abilities. Children with Rett syndrome typically begin to lose the ability to speak, to make eye contact and to communicate in other ways. They may become disinterested in other people, toys and their surroundings. Some children have rapid changes, such as a sudden loss of language. Over time, children may gradually regain eye contact and develop nonverbal communication skills.
- Unusual hand movements.
 Children with Rett syndrome usually develop repetitive, purposeless hand movements, which differ from child to child. Hand movements may include hand-wringing, squeezing, clapping, tapping or rubbing.
Children with Rett syndrome usually develop repetitive, purposeless hand movements, which differ from child to child. Hand movements may include hand-wringing, squeezing, clapping, tapping or rubbing.
Other signs and symptoms can include:
- Unusual eye movements. Children with Rett syndrome tend to have unusual eye movements, such as intense staring, blinking, crossed eyes or closing one eye at a time.
- Breathing problems. These include breath holding, rapid breathing (hyperventilation), forcefully blowing out air or saliva, and swallowing air. These problems tend to occur during waking hours. Other breathing disturbances such as shallow breathing or short periods of stopping breathing (apnea) can occur during sleep.
- Irritability and crying. Children with Rett syndrome may become increasingly agitated and irritable as they get older. Periods of crying or screaming may begin suddenly, for no apparent reason, and last for hours.
 Some children may experience fears and anxiety.
Some children may experience fears and anxiety. - Other unusual behaviors. These may include, for example, sudden, odd facial expressions and long bouts of laughter, hand licking, and grasping of hair or clothing.
- Intellectual disabilities. Loss of skills may be connected to losing the ability to think, understand and learn.
- Seizures. Most people who have Rett syndrome experience seizures at some time during their lives. Multiple seizure types may occur and are associated with changes on an electroencephalogram (EEG).
- Sideways curvature of the spine (scoliosis). Scoliosis is common with Rett syndrome. It typically begins between 8 and 11 years of age and progresses with age. Surgery may be required if the curvature is severe.
- Irregular heartbeat. This is a life-threatening problem for many children and adults with Rett syndrome and can result in sudden death.
- Sleep disturbances. Problems with sleep patterns can include irregular sleep times, falling asleep during the day and being awake at night, or waking in the night with crying or screaming.
- Other symptoms. A variety of other symptoms can occur, such as a decreased response to pain; small hands and feet that are usually cold; problems with chewing and swallowing; problems with bowel function; and teeth grinding.
Stages of Rett syndrome
Rett syndrome is commonly divided into four stages:
- Stage 1: Early onset. Signs and symptoms are subtle and easily overlooked during the first stage, which starts between 6 and 18 months of age. Stage 1 can last for a few months or a year. Babies in this stage may show less eye contact and start to lose interest in toys. They may also have delays in sitting or crawling.
- Stage 2: Rapid deterioration. Starting between 1 and 4 years of age, children lose the ability to perform skills they previously had.
 This loss can be rapid or more gradual, occurring over weeks or months. Symptoms of Rett syndrome occur, such as slowed head growth, abnormal hand movements, hyperventilating, screaming or crying for no apparent reason, problems with movement and coordination, and a loss of social interaction and communication.
This loss can be rapid or more gradual, occurring over weeks or months. Symptoms of Rett syndrome occur, such as slowed head growth, abnormal hand movements, hyperventilating, screaming or crying for no apparent reason, problems with movement and coordination, and a loss of social interaction and communication. - Stage 3: Plateau. The third stage usually begins between the ages of 2 and 10 years, and it can last for many years. Although problems with movement continue, behavior may slightly improve, with less crying and irritability, and there may be some improvement in hand use and communication. Seizures may begin in this stage and generally don't occur before the age of 2.
- Stage 4: Late motor deterioration. This stage usually begins after the age of 10 and can last for years or decades. It's marked by reduced mobility, muscle weakness, joint contractures and scoliosis. Understanding, communication and hand skills generally remain stable or improve slightly, and seizures may occur less often.

When to see a doctor
Signs and symptoms of Rett syndrome can be subtle in the early stages. See your child's health care provider right away if you begin to notice physical problems or changes in behavior after what appears to be typical development. Problems or changes may include:
- Slowed growth of your child's head or other parts of the body
- Decreased coordination or mobility
- Repetitive hand movements
- Decreasing eye contact or loss of interest in usual play
- Delayed language development or loss of previous language abilities
- Any clear loss of previously gained milestones or skills
Request an Appointment at Mayo Clinic
Causes
Rett syndrome is a rare genetic disorder. Classic Rett syndrome, as well as several variants (atypical Rett syndrome) with milder or more-severe symptoms, occur based on several specific genetic changes (mutations).
The genetic changes that cause Rett syndrome occur randomly, usually in the MECP2 gene. Very few cases of this genetic disorder are inherited. The genetic changes appear to result in problems with the protein production critical for brain development. However, the exact cause is not fully understood and is still being studied.
Very few cases of this genetic disorder are inherited. The genetic changes appear to result in problems with the protein production critical for brain development. However, the exact cause is not fully understood and is still being studied.
Rett syndrome in males
Because males have a different chromosome combination from females, males who have the genetic changes that cause Rett syndrome are affected in devastating ways. Most of them die before birth or in early infancy.
A very small number of males have a different genetic change that results in a less destructive form of Rett syndrome. Similar to females with Rett syndrome, these males are likely to live to adulthood, but they're still at risk of a number of intellectual and developmental problems.
Risk factors
Rett syndrome is rare. The genetic changes known to cause the disease are random, and no risk factors have been identified. In a very small number of cases, inherited factors — for instance, having close family members with Rett syndrome — may play a role.
Complications
Complications of Rett syndrome include:
- Sleep problems that cause significant sleep disruption to the person with Rett syndrome and family members.
- Difficulty eating, leading to poor nutrition and delayed growth.
- Bowel and bladder problems, such as constipation, gastroesophageal reflux disease (GERD), bowel or urinary incontinence, and gallbladder disease.
- Pain that may accompany problems such as gastrointestinal issues or bone fractures.
- Muscle, bone and joint problems.
- Anxiety and problem behavior that may hinder social functioning.
- Needing lifelong care and assistance with activities of daily living.
- Shortened life span. Although most people with Rett syndrome live into adulthood, they may not live as long as the average person because of heart problems and other health complications.
Prevention
There's no known way to prevent Rett syndrome. In most cases, the genetic changes that cause the disorder occur spontaneously. Even so, if you have a child or other family member with Rett syndrome, you may want to ask your health care provider about genetic testing and genetic counseling.
In most cases, the genetic changes that cause the disorder occur spontaneously. Even so, if you have a child or other family member with Rett syndrome, you may want to ask your health care provider about genetic testing and genetic counseling.
By Mayo Clinic Staff
Related
Associated Procedures
Products & Services
Rett syndrome - treatment, symptoms, causes of the disease, first signs
Rett syndrome - treatment, symptoms, causes of the disease, first signsHome
Disease reference
Retta
Description
- Description
- Symptoms
- Diagnostics
- Treatment
- Who treats
- where to treat
Published: August 15, 2021Time to read: 8 minutesComments
Content author
Description
Rett syndrome is a genetic neuropsychiatric disease that leads to total dementia and loss of motor skills. A synonymous name is cerebroatrophic hyperammonemia. ICD-10 code - F84.2.
A synonymous name is cerebroatrophic hyperammonemia. ICD-10 code - F84.2.
The syndrome is named after the Austrian neurologist and pediatrician Andreas Rett. In 1954, he reported a new and unique condition in several girls. His characteristic symptoms were a regression of mental development and stereotypical hand movements such as "rubbing" or "washing" the palms. Over the next 12 years, A. Rhett collected clinical data on similar cases in Europe, keeping video recordings of the behavior of patients and medical records. His publications received little attention. As an independent disease, Rett syndrome was recognized only in 1983 thanks to the work of Bengt Hagberg and Jean Aicardi.
For a long time it was believed that Rett syndrome belongs to the group of progressive neurodegenerative pathologies. But now most researchers agree that this disease is a genetically determined violation of the development of the brain. The prevalence is 1 case per 10-15 thousand newborn girls. Among boys, pathology is extremely rare. Until 1990, no cases of Rett syndrome were reported among males.
Among boys, pathology is extremely rare. Until 1990, no cases of Rett syndrome were reported among males.
As a rule, random mutations are the cause of the disease. Less than 1% of patients have a hereditary variant of the syndrome. The exact mechanism of the occurrence of the mutation has not been established, but since the gene anomaly is fixed in the X chromosome of the father, who does not have signs of the disease, it presumably occurs during the period of spermatogenesis. Girls with Rett syndrome are born full-term, as a result of pregnancies without complications (80% of cases).
The pathogenesis of the disease is not well understood. It has been established that the brain of sick children is reduced and weighs 850 grams. This is probably due to insufficient branching of dendrites (short processes of neurons), crowding, and small cell size. There are no signs of neurodegeneration. Violation in the development of the brain leads to changes in the structure of dendrites, synaptic contacts of pyramidal cells are not formed in the required volume or undergo reduction.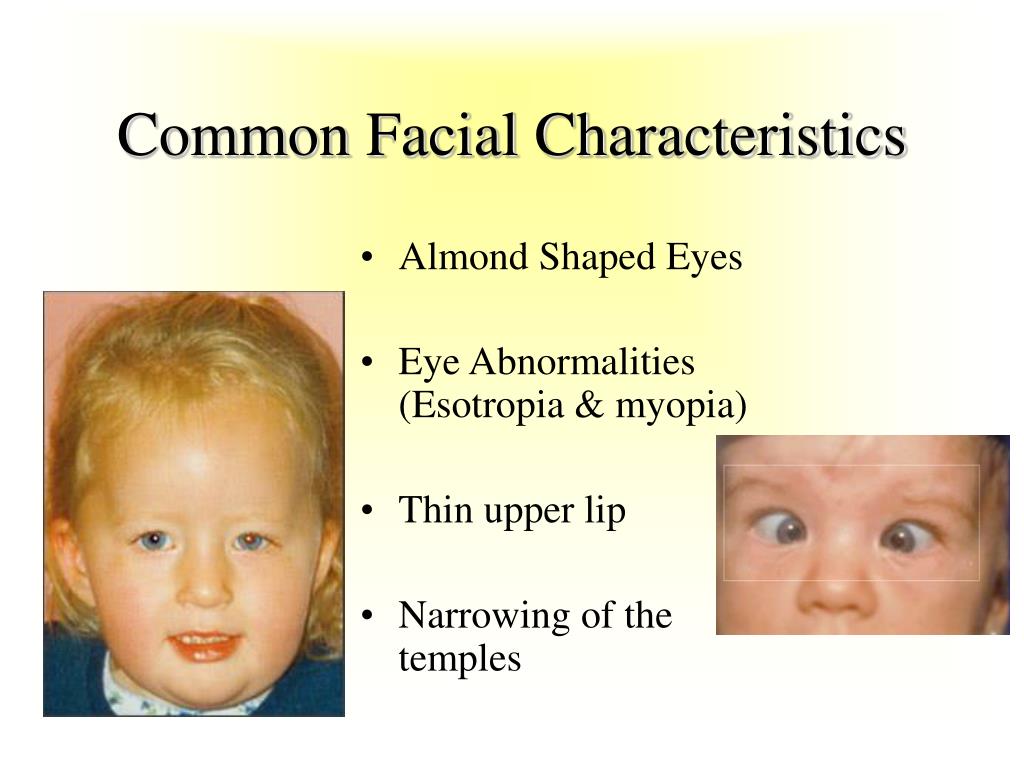 As a result, normal postnatal maturation of the brain is impossible.
As a result, normal postnatal maturation of the brain is impossible.
Symptoms
Photo: doktor-ok.com
In the first six months of life, the development of the child does not differ from normal, with the exception of a few mild symptoms that parents may not pay attention to, and doctors consider as a reason for further observation. These manifestations include reduced muscle tone and a slight lag in the formation of crawling, walking, and sitting skills. Noticeable deviations in development are found in the period from 4-6 months to 2.5 years. The disease progresses, its 4 stages are distinguished:
- Stagnation. Lasts up to 1.5-2.5 years. During this period, delayed psychomotor development is observed. The beginning of cooing and the formation of speech (pronunciation of individual words and simple sentences) are late. The child later begins to understand the requests of the parent. Motor skills are lagging behind, for example, shifting a toy from one hand to another.
 Muscle tone is diffusely reduced. Insufficient growth rate of the head. Gradually, interest in games, in the appeals of mother and other close people, decreases.
Muscle tone is diffusely reduced. Insufficient growth rate of the head. Gradually, interest in games, in the appeals of mother and other close people, decreases. - Regression. Begins at 1-3 years old, ends by preschool age. episodes of inexplicable anxiety, an inconsolable cry, sleep disturbances appear. In a few weeks, all acquired motor skills are lost, speech breaks up. Purposeful hand movements are replaced by stereotypes, reminiscent of the process of washing hands and fingers. Half of sick children suffer from respiratory disorders in the form of apnea (delays), followed by episodes of hyperventilation. Sometimes seizures develop as in epilepsy. Loss of contact with other people makes Rett syndrome similar to autism.
- Stabilization. The third stage continues throughout the preschool and primary school period. The relative stability of the state is observed. All signs of deep mental retardation are determined. Seizures occur at regular intervals. On the part of the motor sphere - hyperkinesis, ataxia, muscle weakness.
 The emotional state improves: a calm sleep is established, episodes of anxiety and screaming pass, it is possible to establish contact with the child.
The emotional state improves: a calm sleep is established, episodes of anxiety and screaming pass, it is possible to establish contact with the child. - Progression of movement disorders. Stage starts at 9-11 years. Immobility develops, spastic changes in muscles and atrophy increase. There are secondary deformations of the bones of the skeleton, such as scoliosis, kyphosis. Pathological changes in the tone of blood vessels are most noticeable in the legs, manifested by a violation of thermoregulation. Children are stunted, but puberty is age appropriate. Perhaps a state of exhaustion of the body (cachexia). Seizures develop less frequently. It becomes possible to communicate at the level of emotions, as well as with the help of pictures, tables and other symbolic material.
The division of Rett syndrome symptoms into 4 stages is conditional. In practice, it is impossible to distinguish between transitions, the disease is characterized by a progressive course.
Diagnosis
Photo: detmed. ru
ru
Children are examined by neurologists and psychiatrists. The diagnosis is established on the basis of clinical signs. Criteria for diagnosis are divided into three groups. Group I - necessary criteria. All of them are present in a patient with the classic form of the syndrome. Group II describes signs that are additional. They are observed in many patients, but are not required to confirm the diagnosis. Group III includes exclusion criteria: if at least one of them is present, then Rett syndrome is rejected.
Required criteria include:
- normal pregnancy and child development in the first 7 days of life;
- head circumference is normal immediately after birth, its growth slows down between 5 months and 4 years;
- purposeful hand movements are lost between 6 and 30 months of age, at the same time the ability to communicate is impaired;
- gross lag in psychomotor development, impaired expressive (pronunciation) and impressive (understanding) speech;
- after the loss of purposefulness of hand movements, motor stereotypes arise, similar to squeezing, squeezing, rubbing;
- from 1 to 4 years of age, gait disturbances (unsteadiness, instability, loss of balance) are manifested.

Additional criteria for Rett syndrome are:
- respiratory disorders such as episodes of daytime apnea, hyperventilation episodes, occasional exhalation of air and saliva;
- seizures;
- atrophic muscle changes, spasticity and dystonia;
- disorders of vascular regulation in the periphery;
- scoliotic changes in posture;
- stunting;
- reduced foot size;
- changes in EEG results.
EEG is one of the few informative instrumental studies in Rett syndrome. The background rhythm becomes slow in the waking state, epileptiform discharges are present, sometimes their increase in sleep is noted, regardless of the presence of convulsions. Changes in the encephalogram are noted from about 2 years. At the age of 6-7 years, a monotonous alpha rhythm begins to dominate, which after the 20th anniversary is localized in the parietal zone.
If at least one exclusionary feature is found, then Rett syndrome is not confirmed. This group of criteria includes:
This group of criteria includes:
- delayed fetal growth;
- manifestations of lysosomal storage diseases, organ enlargement;
- retinopathy and optic nerve atrophy;
- microcephaly at birth;
- acquired brain injury;
- metabolic and neurological diseases.
Treatment
Photo: mebel-pelikan.ru
There are currently no therapies that can cure a person of Rett syndrome. Medical care is reduced to the use of symptomatic drugs, as well as the use of physiotherapy and psychocorrection. The efforts of specialists are aimed at improving the quality of life of patients, maintaining motor skills and the ability to communicate at the highest possible level. The treatment program includes the following activities:
- Taking anticonvulsants. Patients are prescribed anticonvulsants. They help reduce the frequency of seizures. Melatonin is used to eliminate sleep disorders.
- High-calorie diet. In order to prevent exhaustion, patients are shown a diet high in fat, frequent meals with a break of no more than 3 hours.

- LFK. Therapeutic exercise is necessary to slow down the process of loss of motor skills, in particular walking. The exercises support the flexibility and range of motion of the arms and legs.
- Psychocorrection. Psychological assistance programs are aimed at the formation of a specific way of communication. Children with Rett syndrome are able to understand emotions and gestures, and can be trained to respond by choosing pictures or objects with a specific set value.
Prognosis
Rett syndrome was isolated as a separate disease quite recently, and the possibility of its mass diagnosis occurred even later, so there are not enough clinical data that would allow us to speak with certainty about the prognosis. Some patients die in childhood and adolescence due to epileptic seizures, respiratory disorders, dystrophy and exhaustion. Some patients live up to 20-30 years or even longer. They move in wheelchairs and have partially preserved motor functions or are bedridden. Sometimes there is a reduction of individual symptoms, for example, epileptic seizures.
Sometimes there is a reduction of individual symptoms, for example, epileptic seizures.
The information is for reference only and is not a guide to action. Do not self-medicate. At the first symptoms of the disease, consult a doctor.
Sources
- Kazantseva L.Z. Rett syndrome in children. // Attending doctor. - No. 6. – 1998.
- Bashina V.M., Skvortsov I.A. Rett syndrome and some aspects of its treatment. / Almanac "Healing", 2000. - Issue. 3. - S. 133-138.
- Malinina E. V., Zabozlaeva I. V. . Rett syndrome: diagnostic difficulties (clinico-psychopathological aspects). // Russian Journal of Child Neurology, 2016. - No. 3.
- Mukhin K.Yu., Karpova V.I., Bezrukova I.S., Bolshakova E.S., Mironov M.B., Petrukhin A.S. Rett syndrome (literature review and description of a clinical case) // Russian Journal of Child Neurology. 2010. №2.
- Malvina Nodariyevna Kharabadze, V. Yu. B. Pharmacological correction of mitochondrial disorders in Rett syndrome in children // PF.
 2003. No. 1.
2003. No. 1.
Your comments about symptoms and treatment
If you notice a mistake in the text, please highlight it and press Ctrl+Enter
Found error
Rett Syndrome (SR)
In 1966 Austrian pediatrician Andreas Rett described 2 girls with a similar condition. In 1983, Bengt Hagberg and Jean Aicardi published the first paper on 35 cases in English. In 1984, the formation of IRSA, and in 19In 1985, the first congress of doctors on the problem of Rett syndrome was held.
Rett's syndrome is a progressive hereditary disease that manifests itself in early childhood and is characterized by regression of psychomotor development, autistic features, loss of motor skills and specific motor automatisms, and with the frequent development of epilepsy.
Genetics SR:
- SR is characterized by impaired brain development caused by a mutation of the MECP2 transcription regulator gene encoding Methyl-CpG binding protein-2 on the X chromosome, the Xq28 locus (Schaner et al.
 , 2004 ). 8 major mutations of this gene are known, which are detected in 80% of patients with SR
, 2004 ). 8 major mutations of this gene are known, which are detected in 80% of patients with SR - The MECP2 gene regulates the processes of maturation of the central nervous system. Mutation leads to regression of psychomotor development and mental disorders, in particular autistic behavior (Huppke & Cartner, 2005)
- Most reported cases worldwide are sporadic, less than 1% are familial
SR prevalence:
- 1-3.5 cases per 10,000 female population
- 10% of cases among mentally retarded female patients
- About 30% of cases of progressive dementia with early onset in girls, the second most common (after Down syndrome) hereditary disease with dementia occurring in girls (Aicardi, 1998)
Debut and course of SR:
- Debut of the disease in the interval from 6 to 20 months.
- Stage 1 (stagnation) from 6 months. up to 1.5 years. Psychomotor development stops, head growth slows down
- stage 2 (rapid regression), 1.
 5 to 3 years. Regression with the loss of a purposeful manual praxis and the appearance of manual stereotypes: “washing”, “twisting”, “rubbing”, “hands to mouth”
5 to 3 years. Regression with the loss of a purposeful manual praxis and the appearance of manual stereotypes: “washing”, “twisting”, “rubbing”, “hands to mouth” - Stage 3 (pseudo-stationary). Stabilization of severe mental retardation with autistic behavior
- stage 4 (late movement disorders). After 8 years. Stable severe intellectual disorders. Increasing movement disorders
Rett syndrome criteria (J Aicardi 1998, Percy & Lane 2005)
- Normal perinatal period
- Normal head circumference measurements at birth
- Relatively normal psychomotor development up to 6 months.
- Postnatal head growth retardation from 5 months. up to 4 years
- Loss of purposeful manual activity occurring between 6 and 30 months, associated with impaired communication and loss of social skills
- Loss of speech associated with severe mental retardation
- Appearance of stereotyped manual movements.
 These manifestations occur following the loss of purposeful hand movements
These manifestations occur following the loss of purposeful hand movements - Stereotypic movements in the facial muscles: chewing, tapping teeth, baring teeth
- Onset of trunk ataxia and apraxia of walking at 1-4 years
Epilepsy in SR:
Paroxysmal disorders in SR: hyperventilatory attacks, vaso-vagal syncope and epilepsy (J Aicardi 1998). The debut of epilepsy in SR at the age of 5-15 years.
Characteristics of epileptic seizures in SR:
Type 1 - early form (debut from 10 days to 10 months) with frequent generalized seizures and infantile spasms. In the future, seizures have polymorphism and resistance to therapy.
2 type - late childhood form (debut 1.5-6 years). Rare tonic-clonic seizures and myoclonic, atonic, atypical absences predominate.
Reflex attacks in SR: food provocation, stress, hyperventilation. Often - autoinduction.
Features of the course and prognosis of epilepsy depend on the nature of the mutation in SR:
- Early onset has a severe course of epilepsy.
- Late childhood debut - rare seizures and a high percentage of remission.
- In some favorable cases, after 8-10 years, there is stabilization and a decrease in the frequency of attacks, even without therapy.
Features of the EEG in SR:
Regional and multiregional activity of an acute-slow wave, diffuse peak-wave discharges of a low degree of synchronization, the morphology of the complexes can be of the BEPD type.
Treatment of epilepsy in SR:
- At the onset of infantile spasms, Sabril (Vigabatrin) at an average dose of 100 mg/kg/day
- In atypical absences, myoclonic and atonic seizures - valproates (depakin chrono, depakin chronosphere, convulex retard) in monotherapy 30-60 mg/kg/day. In combination with succinimides (Petnidan syrup) at a dose of 20-35 mg/kg/day
- For focal and predominance of generalized tonic-clonic seizures - Trileptal at a dose of 20-40 mg / kg / day, or Tegretol CR 15-20 mg / kg / day, it is better to use in combination with valproate
Authors: K.