Toxic family syndrome
4 Signs You Grew Up in a Toxic Family and How to Overcome it
Psychological trauma is not always a result of growing up in a toxic family environment, although it can be a contributing factor. A child can experience a car accident resulting in a parent’s death which may cause psychological trauma. On the other hand, someone can grow up in a dysfunctional family, which can do extreme damage and have a lasting impact on their life.
What is a Toxic Family?
Every family has moments of discord when they argue or situations when they unintentionally hurt each other. Some families have a history of conflict or friction during every encounter, leaving family members worn out and frazzled when they are together. If you become the worst version of yourself around your family, you may have been raised in a toxic environment.
A parent’s love should be unconditional, but you may have been given ultimatums that if you don’t do something or act a certain way, you won’t be part of the family. You and a sibling can be completely different people and that should be celebrated. But in a toxic family, the constant comparison can leave you feeling attacked and insecure.
Signs of a Toxic Family
-
The Parents Overreact
When a parent is upset over a family heirloom being broken, it may be justified. But if a parent gets upset over small, seemingly unimportant things all the time, this causes feelings of anxiety and fear. A child never knows when they will get yelled at or what type of behavior is inappropriate since a parent act irrationally and inconsistent over many different things.
-
You Feel Anxious
Many children who grow up in a toxic environment are diagnosed with anxiety disorders. This comes from a lack of security, an unstable environment, or mental and physical mistreatment. Some signs your family is toxic include feeling worried, tense, irritable, or restless. It is difficult to have lasting relationships due to a lack of trust in others or their own low esteem.
-
You Criticize Yourself
The constant demeaning from a destructive parent or sibling causes a child to feel unworthy or undeserving. These signs of a toxic family show up in adulthood when they start criticizing themselves, second-guessing their choices, and hesitating when making decisions. This is difficult to change because they truly feel they are worse than others and have no mental or emotional support to teach them otherwise.
-
They Drain Your Energy
Toxic parents and siblings can leave a child feeling defeated because of their own neediness and the tendency they have to be high maintenance. This can leave a child or an adult drained of any extra energy.
How to Overcome a Toxic Family and When to Get Help
One of the most helpful ways you can change your behavior around your toxic family is to set boundaries. As an adult, you’re not obligated to be with your family all the time. You can decide what works best for you and set limits on the amount of time you visit, where you meet, or what you will discuss with them.
Some people remove themselves completely, but others learn to not react, or they learn not to take responsibility for someone else’s feelings, wants, or needs. They talk about neutral topics and avoid reacting to negative comments to show they are disengaged. When a toxic parent or sibling is unable to trigger a rise out of you, they will usually stop the harassment. But if they don’t, you can always leave and avoid the situation.
Feelings of extreme anxiety, low self-esteem, worthlessness, difficulty trusting others, maintaining close relationships, or feeling worn out after a visit with your family are all signs you grew up in a toxic family. Talking with supportive friends, maintaining positive relationships, taking care of your body by eating, sleeping, and staying hydrated are some ways to help yourself.
If the negative feelings become overwhelming and the psychological trauma has been severe, this is the time to reach out for professional help. There are people who are trained to work with this type of trauma and can help relieve the signs associated with a toxic family.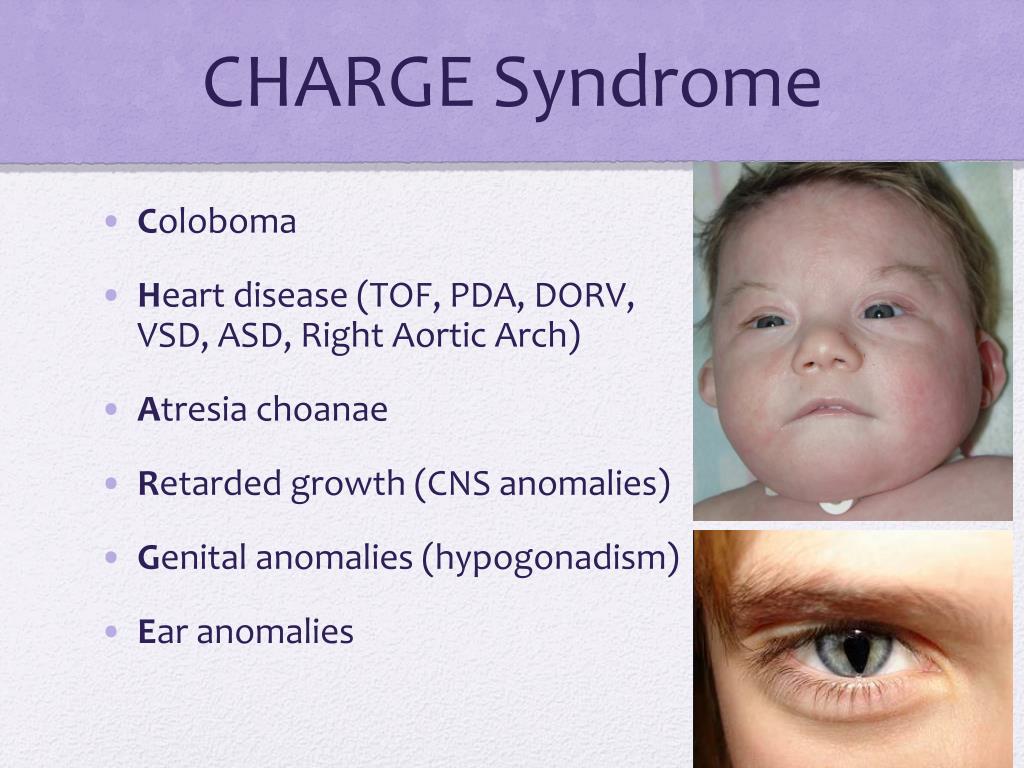
Getting started with treatment for psychological trauma is easy and just a phone call away. At San Antonio Behavioral Healthcare Hospital, aiding those who are experiencing mental health challenges from trauma is our specialty. We understand your unique needs and can develop a customized program for you. Contact us today to learn more about our psychological trauma treatment options.
15 Signs of a Toxic Family Member, and What to Do About Them
Some lucky people are born into families they adore spending time with—their loving mutual bonds make holidays and multi-generational vacations a drama-free joy. But for others, simply seeing an incoming call from a parent triggers an anxiety that dates back to childhood, and they leave family gatherings feeling hurt, angry, or exhausted. Toxic family dynamics can have far-reaching impact on our lives as adults.
And narcissistic parenting isn’t the only type of toxic family relationship. Fern Schumer Chapman, author of Brothers, Sisters, Strangers: Sibling Estrangement and the Road to Reconciliation
, says that this topic isn’t nearly as talked about. “There’s this expectation that siblings will have sustaining relationships for all of their lives,” she says. “So when you say that you don’t, there’s this question of, ‘is there something wrong with you?”’
Fern Schumer Chapman, author of Brothers, Sisters, Strangers: Sibling Estrangement and the Road to Reconciliation
, says that this topic isn’t nearly as talked about. “There’s this expectation that siblings will have sustaining relationships for all of their lives,” she says. “So when you say that you don’t, there’s this question of, ‘is there something wrong with you?”’
The reality can be much more complicated. Chapman adds that typically, a toxic person is the product of a toxic environment themselves—so they often aren’t even aware of their own harmful patterns. “I always joke that if you have one toxic person in your family, you probably have ten,” she says. “Because that’s what was modeled.” Without intervention, it can be perpetuated further by marrying into other people’s dysfunctional families.
Related Story
- Set Your Intention Every Week With Oprah!
Is someone who you're ideally supposed to be close to actually inspiring an instinct to protect yourself? Here are several signs of a toxic family member, and expert advice on dealing with toxic family—because “drink all of the wine” is not a sustainable plan.
No one's known you longer than your family has, which means they've got a rich back catalog of personal failures to draw from when commenting on your life. Their blunt criticism can wound like a physical jab.
"Toxic parents exhibit a chronic lack of empathy towards their children," says Shannon Thomas, trauma therapist and author of Healing from Hidden Abuse. "These behaviors can manifest through biting remarks about appearance, relationship status, mental or physical health, financial struggles, or career challenges."
Even if they insist they're just teasing, those comments may (even subconsciously) be decimating by design. "It's hard to imagine a parent intentionally taking cheap shots at their children, but it happens when they're toxic," Thomas adds.
They give you the silent treatment.Yes, words can hurt—but so can their absence. If they refuse to speak to you for hours (or even days) following an argument, it's a form of manipulation. This is true regardless of the family member.
If they refuse to speak to you for hours (or even days) following an argument, it's a form of manipulation. This is true regardless of the family member.
"Toxic family members are notorious for using silence as a form of punishment and emotional control," says Thomas. "They find power in being pursued for a relationship.”
They lie—or deny.Even when it’s a lie that doesn’t involve or affect you directly, lack of clarity about the truth creates confusion and cultivates a distrust that leaves you wondering what else isn’t true—particularly when it happens repeatedly. “They may even cover a lie with another lie,” says Chapman. Denial may also take the form of (patently false) blanket statements like, “we don’t have secrets in this house.”
They generalize during disagreements.“Specific details can be debated, but vague accusations are a lot harder to dispute,” Chapman explains. The remarks might sound something like, “it never works out,” or “you always do this.”
The remarks might sound something like, “it never works out,” or “you always do this.”
Maybe they flat-out ask you why you can't be more like the brother you've always felt competitive with, or they praise his successes in ways that emphasize where you fall short. Or, they might share something another family member said about you. "Unhealthy parents will pit their children against one another, or against other members of the family," says Thomas. "They set up scenarios where jealousy and resentment can flourish."
They change the subject to turn the tables on you.In an argument, they might deflect attention by bringing up one of your flaws, instead. Chapman offers this example: You tell a loved one you’re concerned about their drug abuse, and they counter with unrelated claims that you’re a bad parent.
Related Stories
- Oprah Opens Up About Overcoming Her Past Traumas
- What Is Trauma Bonding?
- 5 Signs You Were Raised by a Narcissist

It can be extremely painful when you’re trying to share your hurt over a grievance—or even abuse, enacted by them or another family member—only to be left feeling like you hurt them by bringing it up. They may cry or lash out with righteous anger. Or, they may say something like, “Why can’t you let that go?,” effectively minimizing your negative experiences.
They move the goal posts.“Manipulative people often shift the criteria that people have to meet in order to satisfy them,” says Chapman. “It’s very uncomfortable, because just when you think you’ve achieved what they wanted, it’s not good enough.”
They use threats, harsh language, or violence.This may seem like the most obvious sign of a toxic relationship, but not if it's always been normalized as part of your family dynamic. There’s never any situation in which name-calling or physical intimidation and other forms of domestic violence are justified, and if you fear for your safety, help is available.
This can include guilt trips and backhanded compliments, Chapman says, along with nonverbal communication such as rolled eyes and sighs.
They make your business your great-aunt Lydia's business.A blossoming relationship just ended, and though you had no reason to feel embarrassed, you didn't want the whole world to know about your romantic disappointment. Enter your mother, who's spilled your tale as a way to bond (or worse, share a laugh) with someone else.
According to Thomas, it's not uncommon for a toxic family member to breach your confidence. "They'll often share personal information or life struggles with whoever they deem worthy of knowing, with little-to-no regard for how these breaches of trust impact their children's emotional well-being."
They gaslight you.
A term inspired by the 1944 Ingrid Bergman film Gaslight, gaslighting is a type of emotional abuse in which someone causes the victim to doubt their own understanding of reality. “They deny that the abuse is really happening,” says Chapman. “It’s confusing and overwhelming, because all the sudden you’re doubting that what you see and feel is real.”
Examples she offers include a sibling insisting your childhood experiences weren’t as bad as you remember, or a family member point-blank saying something like, “that didn’t happen—you’re making things up, as usual.”
They ignore boundaries.Setting healthy boundaries is crucial in healthy relationships; these can range from “please don’t call me at work” to asking other family members to respect the rules that you set for your kids. If your wishes aren’t being respected by someone who doesn’t think the boundaries apply to them, it can make you feel like you’re not being respected.
A parent, sibling, or other family member may often place blame for anything that’s wrong on someone else—possibly you, included. While their actions or behavior may not be the sole reason for a given issue, regularly refusing to take any accountability is a red flag.
A toxic sibling may "side with" your parent.
In a well-adjusted family dynamic, there's usually no such thing as "taking sides." But when someone learns poor relationship patterns from a parent, they may try to earn that parent's affection by replicating those patterns and thus normalizing harmful behavior.
"Toxic siblings often become a supporter of an equally toxic parent," Thomas says. "They'll use similar critical language as the parent, and shame the targeted sibling regarding areas of life they might be feeling vulnerable about."
Fostering or playing into a competitive dynamic that's meant to make you feel bad is another type of toxic sibling behavior, as is conveniently forgetting your invite to family get-togethers. "Their goal is to send the clear message that you're not included on purpose, and they'll often gloat about what a wonderful event it was," Thomas explains.
"Their goal is to send the clear message that you're not included on purpose, and they'll often gloat about what a wonderful event it was," Thomas explains.
Beware of repeating toxic patterns with others.
You didn’t choose the family you were raised in, but you can make sure you don't invite new toxic influences into your life by assuming the poor ways they treat you are acceptable. "If one or both parents who raised you exhibited significantly unhealthy traits, your ability to assess red flags in the people you meet will be negatively impacted," says Thomas.
"Without true insight on how our family environment created relational blind spots, we run a high risk of repeating toxic patterns from childhood," she continues. "These could include people-pleasing tendencies, difficulty controlling your anger, or being emotionally unavailable in adult relationships." Auditing your relationships' health through self-examination and the assistance of a mental health professional can help you avoid recreating the toxicity.
Before telling a toxic family member how they make you feel, try this.
If you don't feel that their behavior is extreme enough to warrant cutting off contact—or you’re simply not ready to take that extreme step—you may be tempted to call them out, in an effort to break the cycle. Just be sure to manage your expectations of the conversation: Definitely don't assume you'll get an outright apology, or a sudden improvement in your dynamic. In fact, they may wind up pushing your buttons harder than ever.
Related Stories
- 5 Signs You Were Raised by a Narcissist
- Exactly How to Ask for What You Need—and Stay Firm
"The toxic individual will often attempt to bring a heightened level of emotions to the conversation," Thomas says. "On the other side of the spectrum, they might refuse to discuss your concerns." To help keep your conversation even-keeled and on track, Thomas suggests making a list of the person's most hurtful offenses and sticking to your talking points.
Detachment is crucial.
You have no control over someone else's behavior, but you can work on your own reaction to it. When going no-contact isn't an option that you're willing or able to choose, Thomas recommends forging an emotional boundary with what she calls "detached contact."
"Detached contact centers on our ability to be physically present, but not emotionally wounded by the actions of a family member," Thomas explains. "We consciously recognize the psychological games they're playing to get a reaction out of us, but we refuse to engage in the toxicity." Instead, she says, invest your energy in healthier family members who treat you with respect, and "deflect all attempts by the toxic person to engage in an argument or drama." Placing distance between your emotions and their chaos-sowing tactics isn't simple, but it does get easier with practice.
Want more stories to inspire you to live your best life? Sign up to become an Oprah Insider!
When
should you cut them off?Deciding to enforce a no-contact rule is a big move that may test your resolve, call for new family holiday traditions, and spur other family members to try and intervene. It’s certainly not the sole option for every turbulent family bond (see the other possible paths above), nor is it the right option for everyone. It also doesn’t always have to be permanent; in her book, Chapman writes about the long road to successfully repairing her relationship with her own long-estranged brother.
It’s certainly not the sole option for every turbulent family bond (see the other possible paths above), nor is it the right option for everyone. It also doesn’t always have to be permanent; in her book, Chapman writes about the long road to successfully repairing her relationship with her own long-estranged brother.
But as Thomas points out, certain situations require it—especially when previous attempts to improve relations are unsuccessful. No-contact becomes an option to consider if the situation is significantly impacting your mental health. "An increase in symptoms of depression, anxiety, panic disorder, addictions, and mood instability are all signs of necessary distance from a toxic family member," Thomas says.
"It's an intensely painful experience to face the necessity of cutting a family member out of our lives," she continues. "It's a figurative death with complex grief, because the family member is still living but emotionally unsafe. "
"
Another reason people may choose to protect themselves with a no-contact rule is out of fear that their own children will be exposed to the same unacceptable behaviors or outright abuse. As Thomas notes, "Toxic parents frequently become toxic grandparents."
Samantha Vincenty
Senior Staff Writer
Samantha Vincenty is the former senior staff writer at Oprah Daily.
This content is imported from OpenWeb. You may be able to find the same content in another format, or you may be able to find more information, at their web site.
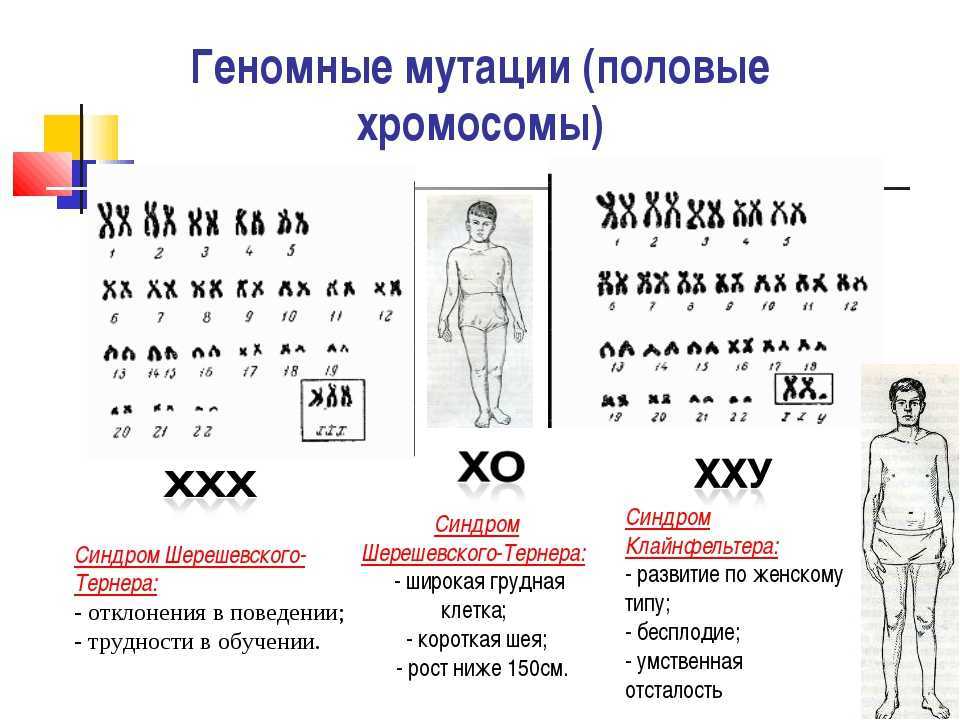
Familial case of hypogonadotropic hypogonadism as a manifestation of CHARGE syndrome | Khabibullina
RELEVANCE
CHARGE syndrome (OMIM no. 214800) is a rare genetic disease with autosomal dominant inheritance, which is based on mutations in the CHD7 gene. The incidence rate ranges from 1:12,000 to 1:15,000 newborns [1][2].
The term "CHARGE" is an acronym for the most commonly encountered clinical features, including coloboma, heart defects, Choanal atresia, and retardation of growth. and/or development), Genitourinary Malformation and Ear abnormalities. nine0007
and/or development), Genitourinary Malformation and Ear abnormalities. nine0007
The first clinical descriptions appear in the literature since 1979, when Hitter et al. and Hall independently described the phenotypic features inherent in the syndrome. In 1981 Pagon et al. [3] proposed to combine the main clinical manifestations into an abbreviation. CHARGE syndrome is currently considered a clinical diagnosis based on a combination of major and minor criteria proposed by Blake et al. in 1998, and a little later expanded and modified by Verloes et al. in 2005 [4]. The authors also proposed to distinguish typical/atypical and partial forms of the syndrome (Table 1). nine0007
Table 1 . Diagnostic criteria of the Charge
| Blake criteria (1998) | Criteria Verloes (2005) | Modified Hale et al. (2016) | |
| The diagnosis is beyond doubt: 4 major criteria Diagnosis possible: 1–2 major criteria and several minor ones | Typical shape: 3 large criteria Atypical: 2 major and no minor, or 1 major + 2 minor. Partial Form: | ChaRGE: 2 large + somewhat small | |
| Large criteria 9000 9000 9000 | |||
| 9000 9000 9000 9000 • Kolobo. nine0007 • Choanal atresia/stenosis. •Hypo- or aplasia of the semicircular canals of the inner ear. • Dysfunction of cranial nerves (mainly VII and VIII pairs ) | • Coloboma. • Choanal atresia/stenosis. •Hypo- or aplasia of the semicircular canals of the inner ear | •Coloboma. • Choanal atresia/stenosis. • Abnormalities of the ear, including hypo- or aplasia of the semicircular canals. • Pathogenic gene variants CHD7 | |
| MINOR CRITERIA | |||
| •Genital hypoplasia. • Developmental delay. • Growth deficit. • Cardiovascular malformations. •Defects of the facial skull. • Tracheoesophageal fistula | • Dysfunction of rhomboencephalic structures of the brain (stem / cranial nerves). • Dysfunction of the hypothalamic-pituitary axis. nine0007 •Malformations of the inner/outer ear. • Abnormalities of internal organs (heart, esophagus). •Intellectual disorders | •Cranial nerve dysfunction. • Structural anomalies of the brain. • Developmental delay/intelligence disorders. • Dysfunction of the hypothalamic-pituitary axis. • Anomalies of the genital and internal organs (heart, esophagus, kidneys) | |

In 2004 Vissers L. E. et al. [5] identified pathological changes in the heterozygous state in gene 9 in patients with CHARGE syndrome0005 CHD7 (OMIM no. 608892), thus establishing the molecular genetic basis of the disease.
E. et al. [5] identified pathological changes in the heterozygous state in gene 9 in patients with CHARGE syndrome0005 CHD7 (OMIM no. 608892), thus establishing the molecular genetic basis of the disease.
Due to the possibility of molecular genetic verification of the diagnosis, descriptions of the CHARGE syndrome associated with mutations in the CHD7 gene, but without a complete clinical picture of the disease, or the so-called atypical mild phenotypes, are increasingly appearing in the literature. Moreover, significant phenotypic variability was noted even among members of the same family with the same pathological substitution. In this connection, given the autosomal dominant type of inheritance, the presence of a pathogenic variant CHD7 in members of the same family in combination with one of the major diagnostic criteria is recommended to be considered sufficient to confirm the CHARGE syndrome, which makes it possible to diagnose patients with an atypical or partial form of pathology [1][5].
In the domestic literature, descriptions of CHARGE syndrome are extremely rare, with a predominant emphasis on the tactics of timely surgical and therapeutic methods for the treatment of congenital malformations, starting from the early neonatal period [6][7]. nine0007
The article presents a clinical description of a familial variant of the syndrome with significant intrafamilial variability of clinical manifestations - from a mild form characterized by an isolated disorder of puberty to a typical form with a full range of clinical manifestations as a result of a defect in the CHD7 gene.
CASE DESCRIPTION
Patient A. was first admitted to the pediatric department of the National Research Center for Endocrinology of the Ministry of Health of Russia at the age of 14 with complaints of delayed sexual development, low growth rates, anosmia. Karyotyping was performed at the place of residence - karyotype 46,XX. A child from the 2nd normal pregnancy, 2 independent urgent births. At birth, body length 54 cm, weight 3270 g. From the anamnesis it is known that the marriage of the parents is not closely related, heredity is burdened: the father has hypoosmia, a history of delayed sexual development (at the age of 25 he received therapy with chorionic gonadotropin (hCG) and 3-month-old a course of replacement therapy with testosterone esters, after which the onset of independent puberty was noted), the final height was 180 cm. My paternal uncle had two fruitless marriages. The proband's brother had a congenital coloboma, left-sided microphthalmia, a horseshoe kidney, at the age of 14 he received combined treatment for germinoma of the left testicle, from the age of 17 he received hormone replacement therapy with testosterone esters. Mother - height 160 cm, menarche at 12.5 years (Fig. 1). Since birth, the patient has been observed at the Eye Microsurgery Center for chorioretinal central coloboma of the eye, involving the optic nerve head (OND), congenital microphthalmos of the right eye.
At birth, body length 54 cm, weight 3270 g. From the anamnesis it is known that the marriage of the parents is not closely related, heredity is burdened: the father has hypoosmia, a history of delayed sexual development (at the age of 25 he received therapy with chorionic gonadotropin (hCG) and 3-month-old a course of replacement therapy with testosterone esters, after which the onset of independent puberty was noted), the final height was 180 cm. My paternal uncle had two fruitless marriages. The proband's brother had a congenital coloboma, left-sided microphthalmia, a horseshoe kidney, at the age of 14 he received combined treatment for germinoma of the left testicle, from the age of 17 he received hormone replacement therapy with testosterone esters. Mother - height 160 cm, menarche at 12.5 years (Fig. 1). Since birth, the patient has been observed at the Eye Microsurgery Center for chorioretinal central coloboma of the eye, involving the optic nerve head (OND), congenital microphthalmos of the right eye. nine0007
nine0007
Figure 1.
Note: in the proband’s sibling (2) — congenital coloboma, left-sided microphthalmia, horseshoe kidney, history of combined treatment for germinoma of the left testicle, from the age of 17 — hormone replacement therapy testosterone; father (3) — hypoosmia, delayed sexual development in anamnesis; paternal uncle (4) had two fruitless marriages (not examined).
On examination: height 152 cm (height SDS: -1.47), weight 46.7 kg (body mass index SDS: 0.2), the phenotype was noteworthy - coloboma, microphthalmia of the right eye, convergent strabismus. Sexual development according to Tanner (B 1, P 1) Ax is absent, Me abs. nine0007
In the hormonal profile, prepubertal basal values of sex hormones were noted: luteinizing hormone (LH) — 0.2 U/l (2.6–12.1), follicle-stimulating hormone (FSH) — 0.66 U/l (1.9 -11.7), estradiol - 53.4 pmol / l (97-592), anti-Mullerian hormone (AMH) - 2.8 ng / ml (0.1-9.85), inhibin B - 18. 7 pg / ml (0-273), prolactin - 73.6 mU / l. To clarify the nature of the delay in sexual development, a test with an analogue of gonadotropin-releasing hormone (GnRH) was performed, according to the results of which the maximum release of LH was 0.9U / l, which indicates in favor of the secondary genesis of hypogonadism (Table 2). However, given the family history, a later onset of independent sexual development is not ruled out.
7 pg / ml (0-273), prolactin - 73.6 mU / l. To clarify the nature of the delay in sexual development, a test with an analogue of gonadotropin-releasing hormone (GnRH) was performed, according to the results of which the maximum release of LH was 0.9U / l, which indicates in favor of the secondary genesis of hypogonadism (Table 2). However, given the family history, a later onset of independent sexual development is not ruled out.
Table 2. Hormonal profile of the patient before a sample with an analogue of gonadotropin-kilizing-hormon and, according to its results,
| lh, units/l | 9000 9000 9000 9000 3 FSH, units/l of the FSH, units/lc. ||
| Baseline | 0.2 | 0.66 |
| Through 60 min* | 1.82 | 3. |
 16
16 Note: LH - luteinizing hormone; FSH is a follicle stimulating hormone.
*— the patient received Buserelin once (intranasally) at 150 mcg (one injection) in each nasal passage. nine0007
Ultrasound of the pelvic organs: the size of the uterus and ovaries corresponded to the age group of 2-7 years. A significant delay in bone age was noted on the radiograph of the hands (CV corresponded to 11.5 years according to the TW20 atlas). An MRI of the brain revealed hypoplasia of the olfactory bulb on the right and aplasia of the olfactory bulb on the left. Taking into account the burdened family history of delayed sexual development, a molecular genetic study was carried out - next generation sequencing using the panel "Hypogonadotropic hypogonadism", where the presence of the heterozygous variant c.619 was revealed3C>T:p.R2065C in the CHD7 gene (NM_017780.4), previously described in the literature for Kalman syndrome (KS) [6]. A similar substitution was found in the father and brother of the proband. Based on the clinical picture, hereditary history, low basal levels of gonadotropins and estradiol, test data with a GnRH analogue in combination with anosmia and hypoplasia of the olfactory bulbs, according to MRI, the diagnosis was made: CHARGE syndrome: coloboma, anosmia, hypogonadotropic hypogonadism. In this connection, a screening was carried out for the presence of other possible components of the disease: data for the presence of pathology of the heart, organs of the urinary system, choanae, and the organ of hearing were not received. nine0007
A similar substitution was found in the father and brother of the proband. Based on the clinical picture, hereditary history, low basal levels of gonadotropins and estradiol, test data with a GnRH analogue in combination with anosmia and hypoplasia of the olfactory bulbs, according to MRI, the diagnosis was made: CHARGE syndrome: coloboma, anosmia, hypogonadotropic hypogonadism. In this connection, a screening was carried out for the presence of other possible components of the disease: data for the presence of pathology of the heart, organs of the urinary system, choanae, and the organ of hearing were not received. nine0007
Given the significant lag in bone age, as well as the presence of a genetically confirmed disease, in order to improve growth prognosis and social adaptation, the patient was recommended hormone replacement therapy with estradiol hemihydrate preparations in minimum starting doses.
DISCUSSION
CHARGE syndrome is a pathological condition caused by mutations in the CHD7 gene (Chromodomain Helicase DNA binding protein 7), also known as chromodomain helicase DNA-binding protein 7. The gene is located on the long arm of the 8th chromosome (8q12) and consists of 38 exons, the first of which is non-coding. nine0007
The gene is located on the long arm of the 8th chromosome (8q12) and consists of 38 exons, the first of which is non-coding. nine0007
During the first 22 days of embryonic development, CHD7 is ubiquitously expressed, regulating the transcription of a number of tissue-specific target genes, the effects of which depend on tissue type and developmental stage. Since the maximum expression of the gene was noted in the undifferentiated neuroepithelium and mesenchyme of the neural crest, many features of the syndrome can be attributed to a violation of the process of cell migration of the latter [8].
Over 500 pathogenic variants have been described to date CHD7 . The most common are nonsense mutations, the second most common are frameshift mutations, splicing mutations are much less common. Missense mutations are more often detected in an isolated variant of hypogonadotropic hypogonadism and are associated with milder phenotypes [9]. With a frameshift or nonsense mutations leading to the formation of a functionally inactive or abnormal protein, a more severe course of the disease is more often noted with involvement of more than one organ in the pathological process. However, an exact correlation between the type of mutation and the severity of clinical manifestations of concomitant defects, even among patients with identical mutations, has not been noted [2][10–12]. nine0007
However, an exact correlation between the type of mutation and the severity of clinical manifestations of concomitant defects, even among patients with identical mutations, has not been noted [2][10–12]. nine0007
The most common endocrine abnormality found in CHARGE syndrome is hypogonadotropic hypogonadism (60–80% of cases). Clinical manifestations of changes in gonadotropic function can vary from late puberty to persistent secondary hypogonadism. In boys, hypogonadism may be suspected if a micropenis is present at birth, often in association with cryptorchidism. In girls, the diagnosis can only be suspected during puberty, in the absence of spontaneous puberty. nine0007
The frequent combination of hypogonadotropic hypogonadism and anosmia due to impaired GnRH neuronal migration is a well-known feature of KS. Since CHD7 is involved in the regulation of gene expression during embryonic development, its influence on the action or expression of ANOS1, FGFR1, FGF8, PROK2 and PROKR2 , which are pathological manifestations of SC, is not excluded. Because KS is one of the phenotypes seen with CHARGE, the authors recommend screening CHD7 in patients with hypogonadism and anosmia, whose diagnosis was not confirmed by molecular genetics [8][9][13].
Because KS is one of the phenotypes seen with CHARGE, the authors recommend screening CHD7 in patients with hypogonadism and anosmia, whose diagnosis was not confirmed by molecular genetics [8][9][13].
To date, there are two cases of hypogonadotropic hypogonadism in the literature, caused by the heterozygous variant c.6193C>T:p.R2065C in the CHD7 (NM_017780.4) gene. Both described patients lacked the complete CHARGE syndrome phenotype. In both cases, there are descriptions of delayed sexual development with impaired sense of smell of varying severity, in combination with phenotypic features associated with atypical and partial forms of the CHARGE syndrome (congenital heart disease, facial skeleton and hearing), as well as mentions of delayed sexual development in members proband families. Thus, the pathogenicity of the mutation is beyond doubt, while confirming the literature data on significant polymorphism of clinical manifestations [9][fourteen].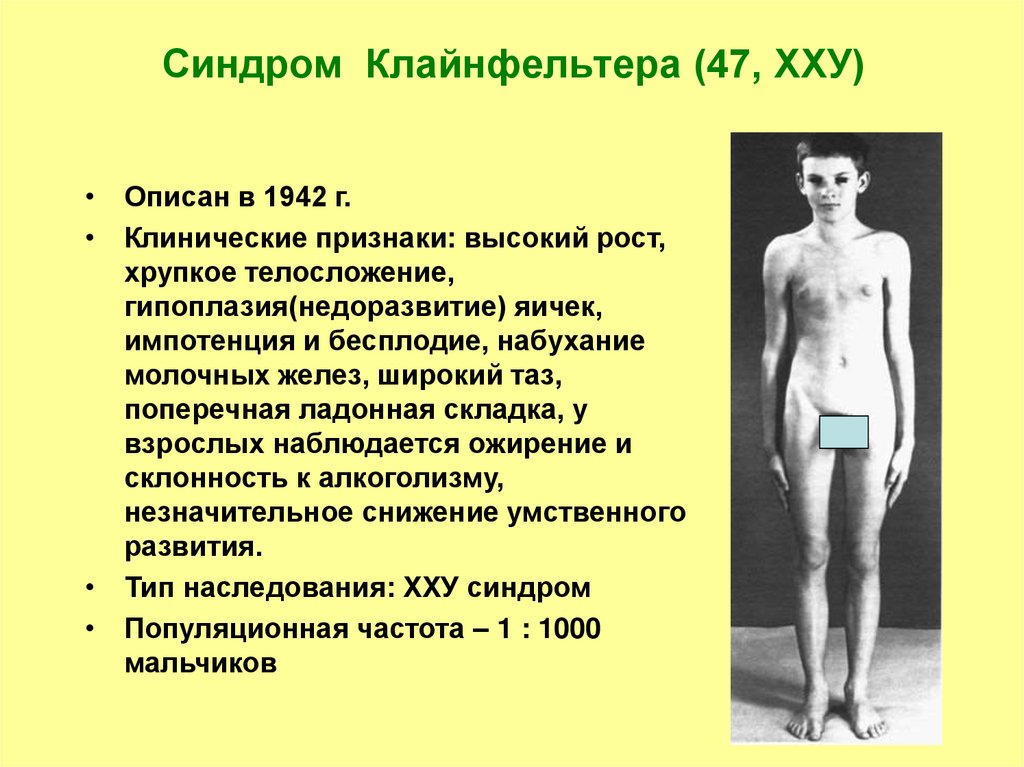
CONCLUSION
Molecular genetic confirmation of the diagnosis is important for genetic counseling and informing patients about all possible clinical manifestations of the disease, in particular about potential reproductive possibilities (in some cases, later independent activation of the hypothalamic-pituitary axis is possible, which does not require hormone replacement therapy or its short-term use), as well as the risks of transmitting the disease to their offspring. nine0007
1. Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet Part A. 2016;170(2):344-354. doi: https://doi.org/10.1002/ajmg.a.37435
2. Chetty M, Roberts TS, Elmubarak M, et al. CHARGE syndrome: genetic aspects and dental challenges, a review and case presentation. Head Face Med. 2020;16(1):10. doi: https://doi.org/10.1186/s13005-020-00224-4
3. Pagon RA, Graham JM, Zonana J, Yong S-L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223-227. doi: https://doi.org/10.1016/S0022-3476(81)80454-4
Pagon RA, Graham JM, Zonana J, Yong S-L. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223-227. doi: https://doi.org/10.1016/S0022-3476(81)80454-4
4. Verloes A. Updated diagnostic criteria for CHARGE syndrome: A proposal. Am J Med Genet Part A. 2005;133A(3):306-308. doi: https://doi.org/10.1002/ajmg.a.30559
5. Vissers LELM, van Ravenswaaij CMA, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955-957. doi: https://doi.org/10.1038/ng1407
6. Leviashvili JG, Savenkova ND, Gorkina OK, et al. CHARGE syndrome. Ross Vestn Perinatol i Pediatr (Russian Bull Perinatol Pediatr). 2020;65(1):116-121. doi: https://doi.org/10.21508/1027-4065-2020-65-1-116-121
7. Garbaruk ES, Nnomzoo AA, Pavlov PV, Gorkina OC. Algorithm of hearing monitoring of children with congenital heart disease. Pediatr (St. Petersburg). 2019;10(2):129-135. doi: https://doi.org/10.17816/PED102129-135
doi: https://doi.org/10.17816/PED102129-135
8. Balasubramanian R, Crowley WF. Reproductive endocrine phenotypes relating to CHD7 mutations in humans. Am J Med Genet Part C Semin Med Genet. 2017;175(4):507-515. doi: https://doi.org/10.1002/ajmg.c.31585
9. Marcos S, Sarfati J, Leroy C, et al. The Prevalence of CHD7 Missense Versus Truncating Mutations Is Higher in Patients With Kallmann Syndrome Than in Typical CHARGE Patients. J Clin Endocrinol Metab. 2014;99(10): E2138-E2143. doi: https://doi.org/10.1210/jc.2014-2110
10. Basson MA, van Ravenswaaij-Arts C. Functional Insights into Chromatin Remodeling from Studies on CHARGE Syndrome. Trends Genet. 2015;31(10):600-611. doi: https://doi.org/10.1016/j.tig.2015.05.009
11. Dijk DR, Bocca G, van Ravenswaaij-Arts CM. Growth in CHARGE syndrome: optimizing care with a multidisciplinary approach. J Multidiscip Healthc. 2019; 12:607-620. doi: https://doi.org/10.2147/JMDH.S175713
12. Moccia A, Srivastava A, Skidmore JM, et al. Genetic analysis of CHARGE syndrome identifies overlapping molecular biology. Genet Med. 2018;20(9):1022-1029. doi: https://doi.org/10.1038/gim.2017.233
Genetic analysis of CHARGE syndrome identifies overlapping molecular biology. Genet Med. 2018;20(9):1022-1029. doi: https://doi.org/10.1038/gim.2017.233
13. Lee YL, Toh L, Yap F. Delayed puberty and anosmia in charge syndrome: A case report. J ASEAN Fed Endocr Soc. 2020;35(1):122-124. doi: https://doi.org/10.15605/jafes.035.01.21
14. Izumi Y, Suzuki E, Kanzaki S, et al. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril. 2014;102(4):1130-1136. doi: https://doi.org/10.1016/j.fertnstert.2014.06.017
Familial case of congenital hyperinsulinism associated with a mutation in the GLUD1 gene | Melikyan
Congenital hyperinsulinism (CHI) is a rare hereditary disease characterized by hypersecretion of insulin by pancreatic beta cells, which leads to the development of hypoglycemia. Late diagnosis and inadequate treatment can lead to severe neurological complications [1]. The incidence of CHI is 1:30,000–1:50,000 live newborns [2]. CHI is an etiologically heterogeneous disease. Currently described 9genes whose mutations can lead to the development of CHI [2]. CHI is most often associated with mutations in the KCNJ11 and ABCC8 genes, which encode proteins that structure ATP-dependent potassium channels in pancreatic beta cells, which, in turn, play a key role in the mechanisms of insulin secretion [2–4].
CHI is an etiologically heterogeneous disease. Currently described 9genes whose mutations can lead to the development of CHI [2]. CHI is most often associated with mutations in the KCNJ11 and ABCC8 genes, which encode proteins that structure ATP-dependent potassium channels in pancreatic beta cells, which, in turn, play a key role in the mechanisms of insulin secretion [2–4].
Activating mutations in the GLUD1 gene are the second most common cause of CHI. Mutations of this gene are detected in 10–15% of cases [5,6]. This form of HHI, due to the clinical picture, is also called hyperammonemia leucine-sensitive hypoglycemia, or the syndrome of hyperinsulinism and hyperammonemia (HI/HA). nine0007
Gene GLUD1 is located on the long arm of chromosome 10 and contains 13 exons encoding the enzyme glutamate dehydrogenase (GDH) consisting of 505 amino acids [7]. GDH is a mitochondrial enzyme that is expressed in high concentrations in the liver, brain, kidneys, pancreas, heart, and lungs [7]. This enzyme catalyses the conversion of glutamate to α-ketoglutarate and ammonium. In beta cells, α-keto glutarate, participating in the Krebs cycle, leads to an increase in the concentration of intracellular ATP. An increase in the ratio of ATP / ADP entails the closure of ATP-dependent K + channels, membrane depolarization and opening of voltage dependent Ca ++ channels, which in turn leads to the penetration of Ca ++ into the cell, stimulating insulin secretion [6]. GDH plays a key role in glutaminolysis and regulation of amino acid-induced insulin secretion [6]. The activity of GDH is regulated by a complex interaction of its allosteric activators and inhibitors. Leucine and ADP have an activating effect on GDH, while GTP is its potential inhibitor [8]. Allosteric activation of glutaminolysis is one of the mechanisms of stimulation of insulin secretion by leucine [7]. nine0007
This enzyme catalyses the conversion of glutamate to α-ketoglutarate and ammonium. In beta cells, α-keto glutarate, participating in the Krebs cycle, leads to an increase in the concentration of intracellular ATP. An increase in the ratio of ATP / ADP entails the closure of ATP-dependent K + channels, membrane depolarization and opening of voltage dependent Ca ++ channels, which in turn leads to the penetration of Ca ++ into the cell, stimulating insulin secretion [6]. GDH plays a key role in glutaminolysis and regulation of amino acid-induced insulin secretion [6]. The activity of GDH is regulated by a complex interaction of its allosteric activators and inhibitors. Leucine and ADP have an activating effect on GDH, while GTP is its potential inhibitor [8]. Allosteric activation of glutaminolysis is one of the mechanisms of stimulation of insulin secretion by leucine [7]. nine0007
Activating mutations in the GLUD1 gene reduce the sensitivity of GDH to its inhibitor GTP and, in rare cases, lead to an increase in the activity of GDH itself [9–11]. The absence of the inhibitory effect of GTP in the presence of the activating effect of leucine leads to hypersecretion of insulin. The pathophysiological mechanism explains the clinical picture of CHI associated with GLUD1 mutations: hypoglycemia develops mainly after a meal containing proteins.
The absence of the inhibitory effect of GTP in the presence of the activating effect of leucine leads to hypersecretion of insulin. The pathophysiological mechanism explains the clinical picture of CHI associated with GLUD1 mutations: hypoglycemia develops mainly after a meal containing proteins.
To date, about 30 different mutations in gene 9 have been described in the literature0005 GLUD1 , most of which are located in exons 11 and 12, encoding the GTP-binding domain of the enzyme [5, 9]. There are also descriptions of mutations in the catalytic domain (exons 6 and 7) [10]. All of them lead to a decrease in the sensitivity of the enzyme to the inhibitory effect of GTP. Recently, descriptions of mutations affecting domain 3 (exon 10) have appeared [10]. Functional studies of these mutations have shown that they increase the activity of the enzyme itself to a greater extent than reduce its sensitivity to the inhibitory effect of GTP [10]. nine0007
Hypoglycemic syndrome in this form of CHI is usually mild and responds well to diazoxide therapy in combination with a low protein diet [5, 6, 8]. Macrosomia at birth is uncommon. The manifestation of hypoglycemic syndrome, as a rule, falls on the second half of life. Hypoglycemia can develop both on an empty stomach and after protein nutrition. Hyperammonemia is a biochemical marker of the GI/HA syndrome and occurs without any clinical manifestations of intoxication (lethargy, headaches, vomiting). Unlike other forms of hyperammonemia in GI/HA, the amino acid profile parameters in patients do not go beyond the normal range [12, 13]. nine0007
Macrosomia at birth is uncommon. The manifestation of hypoglycemic syndrome, as a rule, falls on the second half of life. Hypoglycemia can develop both on an empty stomach and after protein nutrition. Hyperammonemia is a biochemical marker of the GI/HA syndrome and occurs without any clinical manifestations of intoxication (lethargy, headaches, vomiting). Unlike other forms of hyperammonemia in GI/HA, the amino acid profile parameters in patients do not go beyond the normal range [12, 13]. nine0007
R. Kapoor et al. [12] published data on 20 patients with GI/GA syndrome associated with mutations in the GLUD1 gene. All patients were born with normal body weight for gestational age. The mean age of onset of hypoglycemia was 23.4 weeks. Hyperammonemia was laboratory recorded in 19 patients. A good therapeutic effect of diazoxide was observed in more than 90% of patients. The most common clinical symptom was seizures (94%), and 43% of children developed generalized epilepsy. This complication was more often observed in patients with mutations in exons 6 and 7 of gene GLUD1 [12, 14].
Despite the relatively mild course and late onset of the hypoglycemic syndrome, HI/GA syndrome is characterized by generalized epilepsy and severe neurological disorders, the mechanism of which is not completely clear [15]. It is assumed that hyperactivity of GDH in the brain leads to a decrease in the level of glutamine and glutamate, which protect the CNS from neurointoxication [9, 11].
As already noted, CHI associated with mutations in gene GLUD1 , in the vast majority of cases amenable to diazoxide therapy. This drug is a direct agonist of ATP-dependent K + channels of beta cells, which prevents depolarization of the cell membrane and thereby suppresses insulin secretion. Doses of diazoxide in GI/GA syndrome range from 5 to 15 mg/kg/day. In some cases, dietary therapy with partial protein restriction is additionally indicated [16].
Several direct GDH inhibitors have now been described. These include, in particular, catechins - organic substances from the group of flavonoids. Clinical studies of various drugs are being conducted to evaluate their effectiveness in the treatment of HI/HA syndrome [17, 18]. nine0007
Clinical studies of various drugs are being conducted to evaluate their effectiveness in the treatment of HI/HA syndrome [17, 18]. nine0007
We present a clinical description of three patients from the same family (two sisters and their mother) with a genetically verified HI/HA syndrome.
Case description
Patient AND .
Girl (see picture) from a closely related marriage (parents are second cousins), from the second pregnancy, the first urgent surgical delivery. She was born weighing 3700 g, height 54 cm, 8-9 points on the Apgar scale. The neonatal period was uneventful. Up to a year, she grew and developed in accordance with her age. At the age of 1 year in the morning on an empty stomach, the child developed clonic-tonic twitching of the limbs without loss of consciousness. She was consulted by a neurologist at the place of residence, epilepsy was suspected and anticonvulsant therapy (valproic acid) was prescribed. Despite the ongoing treatment, such convulsive attacks persisted up to 3 times a month. nine0007
Despite the ongoing treatment, such convulsive attacks persisted up to 3 times a month. nine0007
Drawing. Pedigree of patients.
Black - the presence of clinical manifestations of the disease; white color - the absence of clinical manifestations of the disease; gray color - presumably the presence of the disease.
At the age of 1 year 3 months, the child was hospitalized at the place of residence, where hypoglycemia (minimally up to 1.4 mmol/l) was recorded for the first time without significant clinical manifestations. The study of the glycemic profile for several days revealed recurrent hypoglycemia (1.9-2.5 mmol/l), which were observed both on an empty stomach and after meals. A fasting test was performed: 10 hours after a meal, subclinical hypoglycemia (2.5 mmol/l) developed, against which the insulin and cortisol values were within the normal range (Table 1) . No abnormalities were found in the spectrum of amino acids and acylcarnitines. An EEG revealed epiactivity in the frontal region. With a diagnosis of hypoglycemia of unspecified genesis, the girl was sent for examination at the Federal State Budgetary Institution ENTS. nine0007
An EEG revealed epiactivity in the frontal region. With a diagnosis of hypoglycemia of unspecified genesis, the girl was sent for examination at the Federal State Budgetary Institution ENTS. nine0007
Table 1 . Fasting results
| Index | Patient AND . | Patient M . |
| Age of the study, months | 15 | 12 |
| Duration of fasting period, h | 10 | 9 |
| Glycemia at the end of the test, mmol/l | 2.5 | 5.07 |
| Ketonuria | Negative | Not investigated |
| Ketonemia, mmol/l | Not investigated | 0. |
| Insulin, mcU/ml | 1.4 | 2.03 |
| Cortisol, nmol/l | 605 | 800 |
 1
1 At the age of 1 year and 8 months, she was first examined at the ERC. Identified recurrent hypoglycemia, mainly in the daytime. Against the background of spontaneously developed hypoglycemia (1.7 mmol/l), an inadequate increase in insulin levels up to 4.4 mcU/ml was detected, ketone bodies were absent in the urine. Based on the results obtained (hypoketotic hypoglycemia, no suppression of insulin secretion), a diagnosis of congenital hyperinsulinism was established and therapy with somatostatin analogs at a dose of 20 μg/kg/day was recommended. During therapy, there was some positive dynamics, but rare episodes of hypoglycemia (up to 2.6 mmol/l) persisted and there were complaints of frequent abdominal pain and diarrhea. Due to side effects, octreotide therapy was discontinued.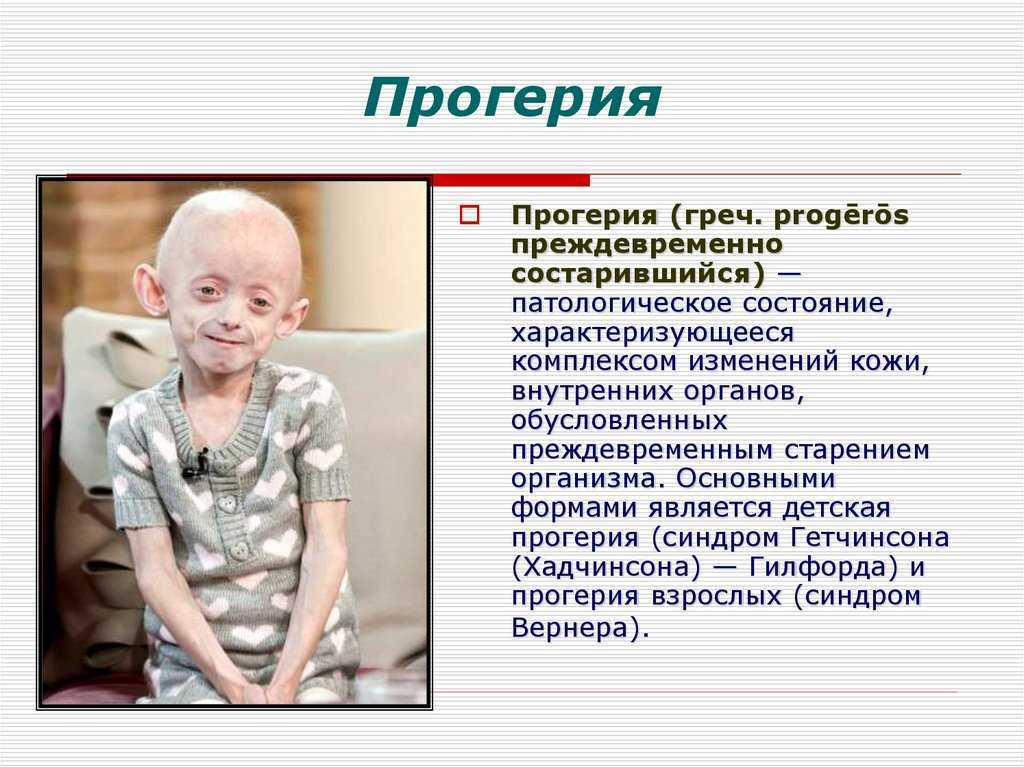 Canceled and anticonvulsants. nine0007
Canceled and anticonvulsants. nine0007
During re-hospitalization at 2 years old, the child was prescribed diazoxide at a dose of 5.5 mg/kg/day, against which stable euglycemia (3.7–6.2 mmol/l) was achieved during the day. The genes KCNJ11, ABCC8 were studied - no mutations were found.
At the age of 3, the child underwent a molecular genetic study of genes GCK, GLUD1 . The previously described heterozygous mutation c.G965A p.R322H (transcript NM_005271) [19] in the GLUD1 gene was identified, which made it possible to verify the diagnosis of protein-induced hypoglycemia. nine0007
The girl is seen annually at the ERC, where the dose of diazoxide is adjusted. At the moment, the child is 7 years old, the dose of diazoxide is 5.56 mg/kg/day. The diet is free, does not adhere to a specific diet. Against the background of taking protein-containing products (meat, eggs, cottage cheese), glycemia indicators are within the normal range. Psychomotor development corresponds to age. Seizures are not noted. Control EEG showed no epiactivity.
Seizures are not noted. Control EEG showed no epiactivity.
Patient A.
Examined at the age of 27 due to a disease in a child (see figure) . A molecular genetic study of the GLUD1 gene revealed an identical heterozygous mutation c.G965A p.R322H. From the anamnesis it is known that until the age of 6 she was observed by a neurologist with a diagnosis of epilepsy and received anticonvulsant therapy. At the age of 6 years, anticonvulsants were discontinued, convulsions did not recur, and the diagnosis was withdrawn. Prior to the discovery of the genetic defect, glycemia was not measured. Didn't follow a specific diet. The mother's sibling died at the age of 1 year for an unspecified reason, the parents are healthy, they were not examined. nine0007
After molecular genetic verification of the diagnosis, an examination was carried out: fasting glycemia was within the normal range, however, after protein loading (a test with a protein mixture of 1 g per 1 kg of body weight), subclinical hyperinsulinemic hypoglycemia was recorded (Table 2) . Against the background of a free diet, hypoglycemia was not recorded. Given the mild course of the disease, diazoxide therapy was not prescribed.
Against the background of a free diet, hypoglycemia was not recorded. Given the mild course of the disease, diazoxide therapy was not prescribed.
Table 2. Results of the oral test with protein loading (1 g per 1 kg of body weight) of patient A ., 27 years old
| Index | 0 ` | 30 ` | 60 ` | 90 ` |
| Glycemia, mmol/l | 5.1 | 4.7 | 3.1 | 2.6 |
| Ketonemia, mmol/l | 0.1 | |||
| Insulin, mcU/ml | 18.9 | 47.2 |
Patient M .
Born at term, body weight 3840 g, height 51 cm, Apgar score 8-9 (see figure) . Due to the presence of molecular genetically confirmed congenital hyperinsulinism in the mother and older sister, the girl in the neonatal period was monitored for glycemia - the indicators were within the normal range. In the future, the mother independently periodically measured glycemia, the minimum values were 2.6 mmol/l; episodes of hypoglycemia proceeded without significant clinical symptoms, were stopped by the intake of sweet water. nine0007
Due to the presence of molecular genetically confirmed congenital hyperinsulinism in the mother and older sister, the girl in the neonatal period was monitored for glycemia - the indicators were within the normal range. In the future, the mother independently periodically measured glycemia, the minimum values were 2.6 mmol/l; episodes of hypoglycemia proceeded without significant clinical symptoms, were stopped by the intake of sweet water. nine0007
At the age of 8 months after breakfast with cottage cheese, hypoglycemia of 2.1 mmol/l was first recorded, accompanied by severe weakness and sweating. Stopped by taking sweet tea. After this episode, protein foods were excluded from the diet. At the age of 1 year, the child was first hospitalized at the Federal State Budgetary Institution ENRC, where a comprehensive examination was carried out, the results of which confirmed the diagnosis of HI/HA syndrome. It should be noted that during the test with starvation, hypoglycemia was not recorded (see Table 1) , whereas in the test with a protein load, hypoglycemia developed after 30 minutes and was accompanied by a pronounced rise in the level of insulin in the blood (Table 3) . A molecular genetic study confirmed the presence of the c.G965A p.R322H mutation in the GLUD1 gene in the girl.
A molecular genetic study confirmed the presence of the c.G965A p.R322H mutation in the GLUD1 gene in the girl.
Table 3 . Results of an oral test with a protein load (1 g per 1 kg of body weight) of the patient M ., 1 year
| Index | 0` | 15` | 30 ` |
| Glycemia, mmol/l | 4.6 | 3.6 | 2.7 |
| Ketonemia, mmol/l | 0.1 | ||
| Insulin, mcU/ml | 6.9 | 37.64 |
The patient was prescribed diazoxide at a dose of 10 mg/kg/day, however, during the control test with protein, subclinical hypoglycemia persisted during therapy, and therefore diet therapy was recommended with restriction of foods high in protein.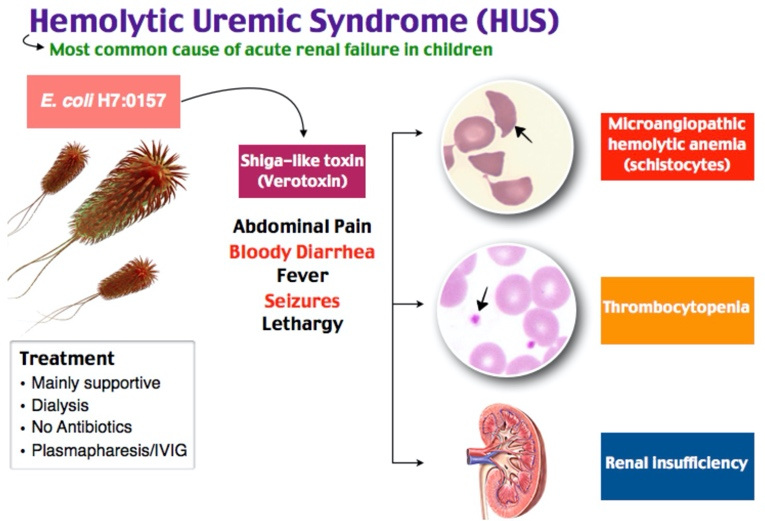
At the age of 2 years, during the next inpatient examination at the ERC, an attempt was made to drink the child with green tea (200-250 ml/day), against which no significant changes in the glycemic profile were observed. When trying to give a protein product (cottage cheese) in combination with green tea, subclinical hypoglycemia (3.1 mmol/l) was recorded after eating. nine0007
The girl is currently 2.5 years old; in a constant mode receives therapy with diazoxide at a dose of 6 mg/kg/day. Against this background, glycemic indices are persistently within the normal range. Diet expanded; the combined intake of protein and carbohydrate products does not lead to the development of hypoglycemia.
Discussion
A familial case of GI/GA syndrome is presented. The diagnosis was confirmed by molecular genetics: all patients had a heterozygous mutation c.G965A p.R322H in exon 7 of gene GLUD1 encoding the catalytic domain of GDH. This mutation has been previously described in the literature.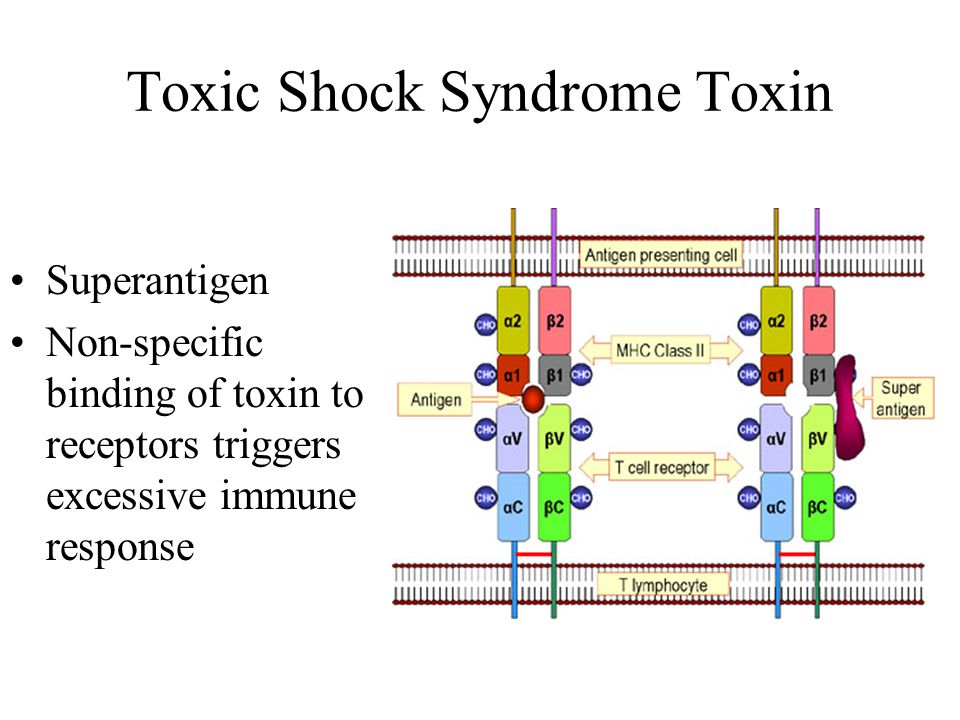 The basal activity of GDH in lymphocytes of patients with this mutation is within the normal range, but the sensitivity of the enzyme to GTP is sharply reduced [19]. According to the literature [12], patients with mutations in exon 7 of GLUD1, are characterized by a milder course of the hypoglycemic syndrome, the absence of severe hyperammonemia, and the frequent development of generalized epilepsy than patients with defects in the allosteric domain (exons 11 and 12) . In our case, a mild course of hypoglycemia was also noted, however, data on epi-activity and neurological deficit in patients were not obtained. nine0007
The basal activity of GDH in lymphocytes of patients with this mutation is within the normal range, but the sensitivity of the enzyme to GTP is sharply reduced [19]. According to the literature [12], patients with mutations in exon 7 of GLUD1, are characterized by a milder course of the hypoglycemic syndrome, the absence of severe hyperammonemia, and the frequent development of generalized epilepsy than patients with defects in the allosteric domain (exons 11 and 12) . In our case, a mild course of hypoglycemia was also noted, however, data on epi-activity and neurological deficit in patients were not obtained. nine0007
The described clinical case is also interesting from the point of view of differential diagnosis. The gold standard for diagnosing CHI is a fasting test and assessment of the level of glycemia, insulin, and ketone bodies [1]. In our case, all 3 patients did not have an increase in insulin levels during the fasting test (see Table 1) , which made it difficult to diagnose and made it possible to doubt the presence of organic hyperinsulinism. However, despite adequate suppression of insulin secretion during fasting, girls (patients and . and M .) there was no increase in ketone bodies. Hormonal verification of the diagnosis was possible only after a test with a load of protein (see Tables 2, 3) . This observation points to the need for an extended diagnostic protocol for patients with hypoketotic hypoglycemias.
However, despite adequate suppression of insulin secretion during fasting, girls (patients and . and M .) there was no increase in ketone bodies. Hormonal verification of the diagnosis was possible only after a test with a load of protein (see Tables 2, 3) . This observation points to the need for an extended diagnostic protocol for patients with hypoketotic hypoglycemias.
In the described family, the parents of the patients M . and AND . were relatives of (see figure) , which led to the initial assumption of an autosomal recessive type of inheritance of the disease, while the HI/GA syndrome is inherited in an autosomal dominant manner. In this situation, sequential analysis of genes (the search for mutations was started from genes KCNJ11 and ABCC8 ) slowed down the molecular genetic verification of the diagnosis. Modern methods of genetic analysis, including parallel sequencing of several genes at once, are preferred in cases of mild forms of CHI.
GI/HA syndrome generally responds well to diazoxide therapy. In our case, the older sister managed to achieve full compensation against the background of this therapy, while the younger girl still needs to restrict dietary proteins during the treatment, and the mother does not need drug therapy at all. Based on the literature data, as well as based on our observations, we can conclude that the course of the disease is milder in adult patients and suggest the possibility of discontinuing drug therapy in girls in the future. nine0007
Indicative is the absence of a significant effect of somatostatin analogs on the level of glycemia in patient AND . This is probably due to the non-selective action of somatostatin and its inability to influence the protein-dependent mechanism of insulin secretion.
Given the data on the inhibitory effect of catechins on GDH, we tried to use green tea (a natural source of these substances) as an additional therapeutic component in a younger girl, but we did not notice significant changes in glycemia.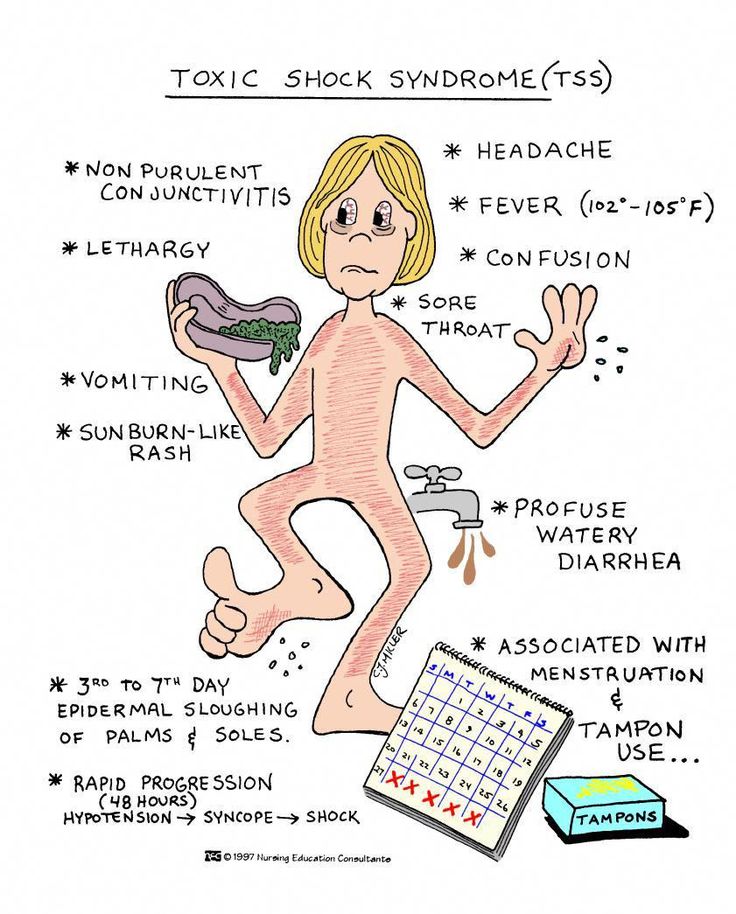 It is possible that higher doses of catechins are needed to suppress the activity of the enzyme. nine0007
It is possible that higher doses of catechins are needed to suppress the activity of the enzyme. nine0007
Conclusion
Clinical and laboratory features of the HI/GA syndrome should be emphasized, characterized by a relatively mild course of the hypoglycemic syndrome and the absence of insulin overproduction on an empty stomach, which necessitates the inclusion of a stimulation test with a protein load in the protocol for examining patients with CHI. Considering the etiological heterogeneity of CHI, the preferred method of molecular genetic diagnosis at the moment is the parallel sequencing of several genes at once (next generation sequencing). Diazoxide is the drug of choice for GI/HA syndrome. nine0007
ADDITIONAL INFORMATION
Consent of the patient. Patients voluntarily signed an informed consent to the publication of personal medical information in anonymized form in the journal "Problems of Endocrinology".
Conflict of interest.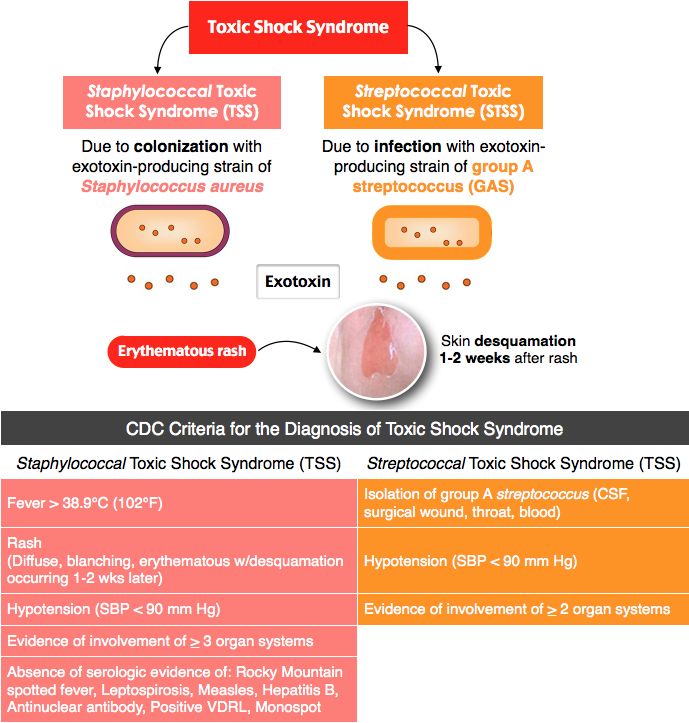 The authors declare no apparent or potential conflicts of interest related to the publication of this article.
The authors declare no apparent or potential conflicts of interest related to the publication of this article.
1. Hussain K, Blankenstein O, De Lonlay P, Christesen HT. Hyperinsulinaemic hypoglycaemia: biochemical basis and the importance of maintaining normoglycaemia during management. Arch DisChild. 2007;92(7):568-570. doi: 10.1136/adc.2006.115543
2. Glaser B, Thornton P, Otonkoski T, Junien C. Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed. 2000;82(2):F79-F86. PMC1721059
3. Thomas P, Cote G, Wohllk N, et al. Mutations in the sulfonylurea receptor gene in familial persistent hyperinsulinemic hypoglycemia of infancy. Science. 1995;268(5209):426-429. doi: 10.1126/science.7716548
4. Thomas P. Mutation of the pancreatic islet inward rectifier Kir6.2 also leads to familial persistent hyperinsulinemic hypoglycemia of infancy. Hum Mol Genet. nineteen96;5(11):1809-1812. doi: 10.1093/hmg/5.11.1809
5. Stanley CA, Lieu YK, Hsu BY, et al.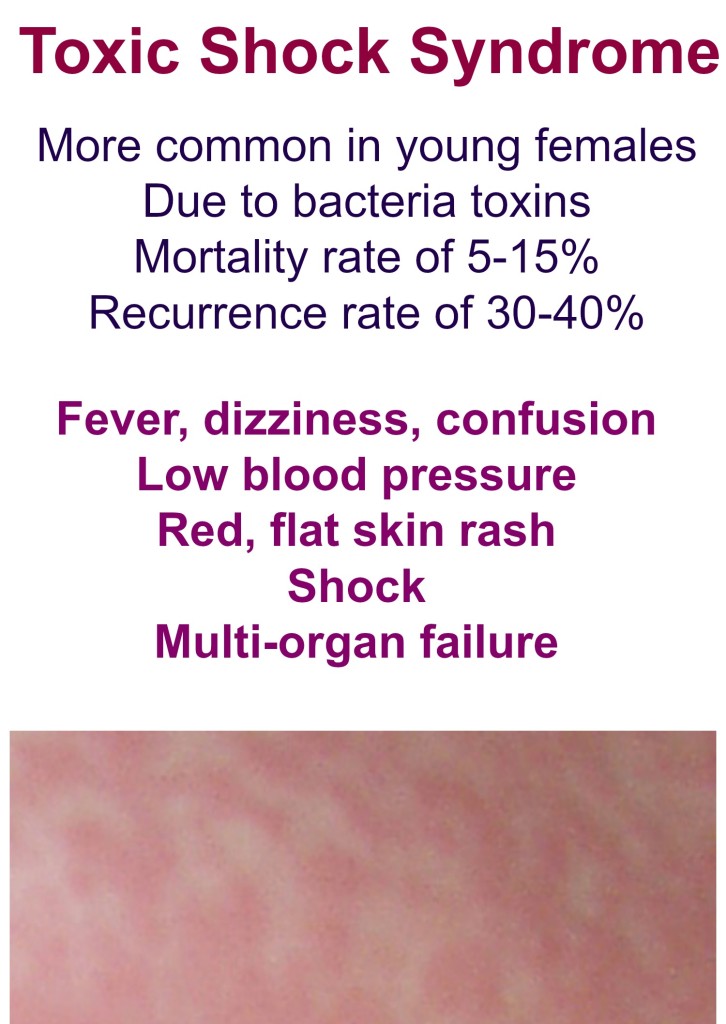 Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338(19):1352-1357. doi: 10.1056/NEJM199805073381904
Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med. 1998;338(19):1352-1357. doi: 10.1056/NEJM199805073381904
6. Weinzimer SA, Stanley CA, Berry GT, et al. A syndrome of congenital hyperinsulinism and hyperammonemia. J Pediatr. 1997;130(4):661-664. doi: 10.1016/s0022-3476(97)70256-7
7. Hudson RC, Daniel RM. l-glutamate dehydrogenases: Distribution, properties and mechanism. Comparative Biochemistry and Physiology Part B: Comparative Biochemistry. nineteen93;106(4):767-792. doi: 10.1016/0305-0491(93)
8. MacMullen C, Fang J, Hsu BYL, et al. Hyperinsulinism/Hyperammonemia Syndrome in Children with Regulatory Mutations in the Inhibitory Guanosine Triphosphate-Binding Domain of Glutamate Dehydrogenase 1. J Clin Endocr Metab. 2001;86(4):1782-1787. doi: 10.1210/jcem.86.4.7414
9. Stanley CA, Fang J, Kutyna K, et al. Molecular basis and characterization of the hyperinsulinism/hyperammonemia syndrome: predominance of mutations in exons 11 and 12 of the glutamate dehydrogenase gene. HI/HA Contributing Investigators. diabetes. 2000;49(4):667-673. doi: 10.2337/diabetes.49.4.667
HI/HA Contributing Investigators. diabetes. 2000;49(4):667-673. doi: 10.2337/diabetes.49.4.667
10. Yorifuji T, Muroi J, Uematsu A, et al. Hyperinsulinism-hyperammonemia syndrome caused by mutant glutamate dehydrogenase accompanied by novel enzyme kinetics. Hum Genet. 1999;104(6):476-479. doi: 10.1007/s0043
11. Stanley CA. Hyperinsulinism/hyperammonemia syndrome: insights into the regulatory role of glutamate dehydrogenase in ammonia metabolism. Mol Genet Metab. 2004;81:Suppl 1:S45-S51. doi:10.1016/j.ymgme.2003.10.013
12. Kapoor RR, Flanagan SE, Fulton P, et al. Hyperinsulinism-hyperammonaemia syndrome: novel mutations in the GLUD1 gene and genotype-phenotype correlations. Eur J Endocrinol. 2009;161(5):731-735. doi: 10.1530/EJE-09-0615
13. Kapoor RR, James C, Hussain K. Advances in the diagnosis and management of hyperinsulinemic hypoglycemia. Nat Clin Pract Endocrinol Metab. 2009;5(2):101-112. doi: 10.1038/ncpendmet1046
14. Bahi-Buisson N, El Sabbagh S, Soufflet C, et al.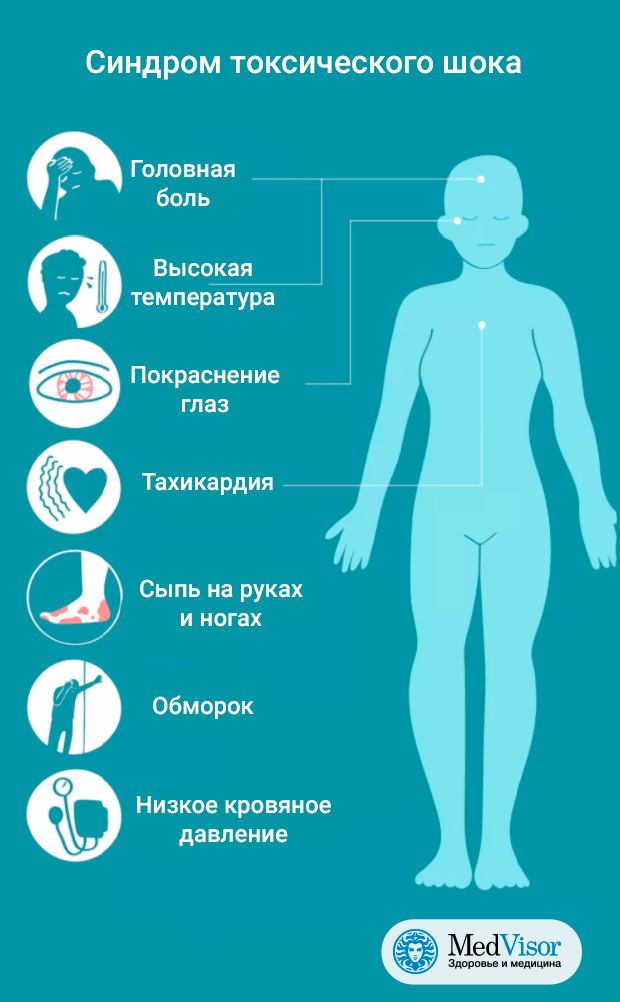 Myoclonic absence epilepsy with photosensitivity and a gain of function mutation in glutamate dehydrogenase. Seizure. 2008;17(7):658-664. doi: 10.1016/j.seizure.2008.01.005
Myoclonic absence epilepsy with photosensitivity and a gain of function mutation in glutamate dehydrogenase. Seizure. 2008;17(7):658-664. doi: 10.1016/j.seizure.2008.01.005
15. Bahi-Buisson N, Roze E, Dionisi C, et al. Neurological aspects of hyperinsulinism-hyperammonaemia syndrome. Dev Med Child Neurol. 2008;50(12):945-949. doi: 10.1111/j.1469-8749.2008.03114.x
16. Palladino AA, Stanley CA. The hyperinsulinism/hyperammonemia syndrome. Rev Endocr Metab Discord. 2010;11(3):171-178. doi: 10.1007/s11154-010-9146-0
17. Li C, Li M, Chen P, et al. Green tea polyphenols control dysregulated glutamate dehydrogenase in transgenic mice by hijacking the ADP activation site. J Biol Chem. 2011;286(39):34164-34174. doi: 10.1074/jbc.M111.268599
18. Li M, Smith CJ, Walker MT, Smith TJ. Novel inhibitors complexed with glutamate dehydrogenase: allosteric regulation by control of protein dynamics. J Biol Chem. 2009;284(34):22988-23000. doi: 10.1074/jbc.M109.020222
19.













![]()
Copyright © · The Wright Initiative · Articles · Site Map
