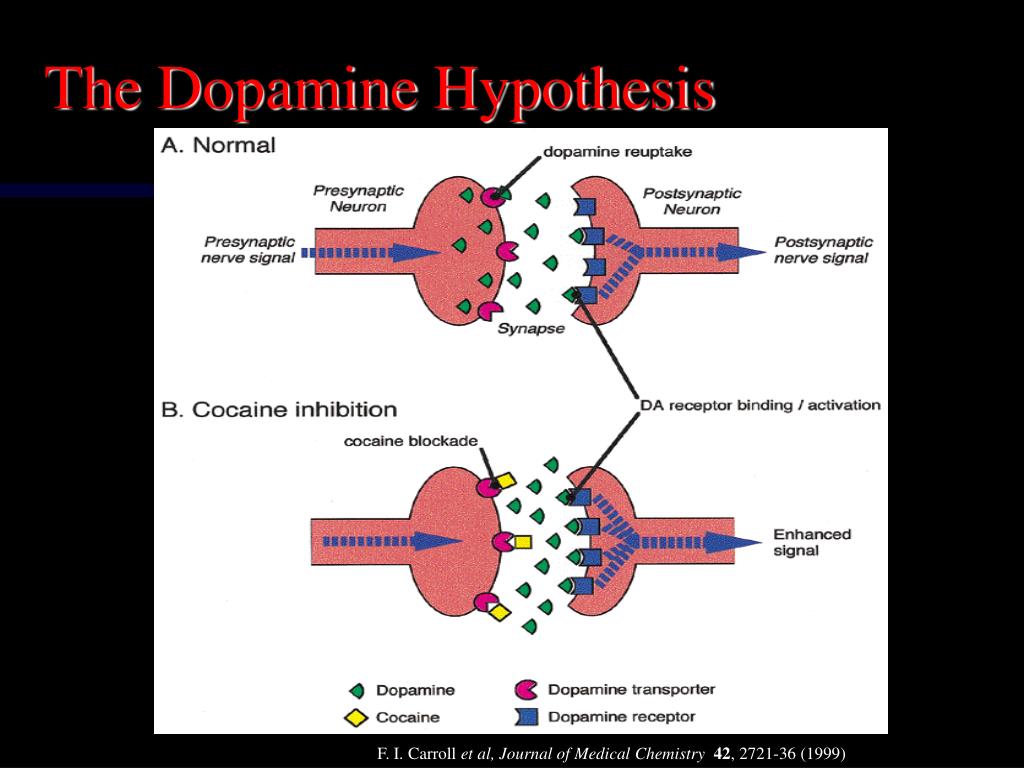
The dopamine hypothesis
The Dopamine Hypothesis of Schizophrenia: Version III—The Final Common Pathway
1. Delay J, Deniker P, Harl JM. Therapeutic use in psychiatry of phenothiazine of central elective action (4560 RP) Ann Med Psychol (Paris) 1952;110:112–117. [PubMed] [Google Scholar]
2. Carlsson A, Lindqvist M. Effect of chlorpromazine or haloperidol on the formation of 3-methoxytyramine and normetanephrine in mouse brain. Acta Pharmacol Toxicol (Copenh) 1963;20:140–144. [PubMed] [Google Scholar]
3. Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. [PubMed] [Google Scholar]
4. Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl) 1987;91:415–433. [PubMed] [Google Scholar]
5. Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–1219. [PubMed] [Google Scholar]
6. Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. [PubMed] [Google Scholar]
7. Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–719. [PubMed] [Google Scholar]
8. Matthysse S. Antipsychotic drug actions: a clue to the neuropathology of schizophrenia? Fed Proc. 1973;32:200–205. [PubMed] [Google Scholar]
9. Snyder SH. The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. Am J Psychiatry. 1976;133:197–202. [PubMed] [Google Scholar]
10. Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–1486. [PubMed] [Google Scholar]
11. Pycock CJ, Kerwin RW, Carter CJ. Effect of lesion of cortical dopamine terminals on subcortical dopamine receptors in rats. Nature. 1980;286:74–76.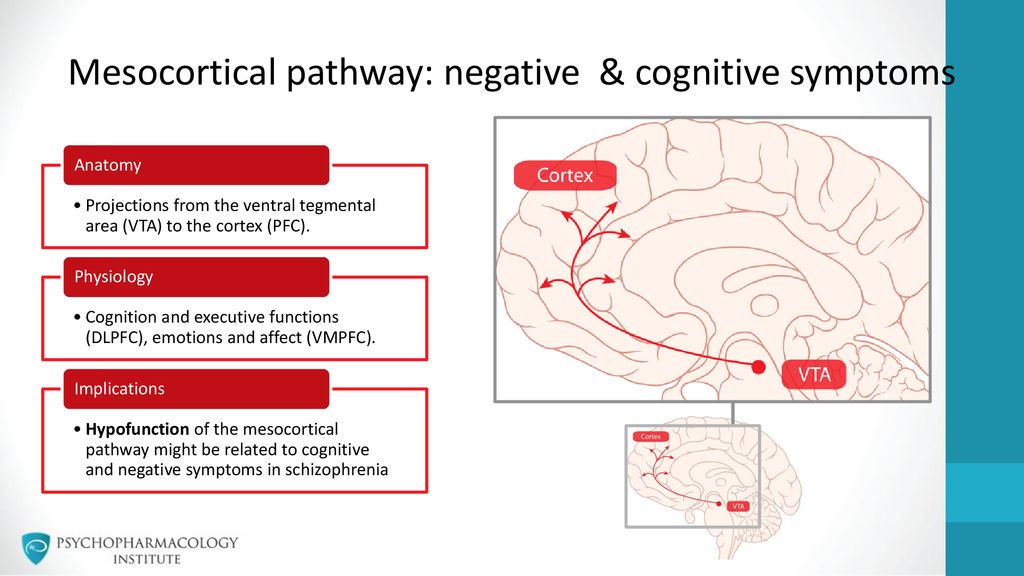 [PubMed] [Google Scholar]
[PubMed] [Google Scholar]
12. Scatton B, Worms P, Lloyd KG, Bartholini G. Cortical modulation of striatal function. Brain Res. 1982;232:331–343. [PubMed] [Google Scholar]
13. Davidson LL, Heinrichs RW. Quantification of frontal and temporal lobe brain-imaging findings in schizophrenia: a meta-analysis. Psychiatry Res. 2003;122:69–87. [PubMed] [Google Scholar]
14. McGuire P, Howes OD, Stone J, Fusar-Poli P. Functional neuroimaging in schizophrenia: diagnosis and drug discovery. Trends Pharmacol Sci. 2008;29:91–98. [PubMed] [Google Scholar]
15. Moore RY, Whone AL, McGowan S, Brooks DJ. Monoamine neuron innervation of the normal human brain: an 18F-DOPA PET study. Brain Res. 2003;982:137–145. [PubMed] [Google Scholar]
16. Meyer-Lindenberg A, Miletich RS, Kohn PD, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267–271. [PubMed] [Google Scholar]
17. McGowan S, Lawrence AD, Sales T, Quested D, Grasby P. Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry. 2004;61:134–142. [PubMed] [Google Scholar]
Presynaptic dopaminergic dysfunction in schizophrenia: a positron emission tomographic [18F]fluorodopa study. Arch Gen Psychiatry. 2004;61:134–142. [PubMed] [Google Scholar]
18. Hietala J, Syvalahti E, Vuorio K, et al. Presynaptic dopamine function in striatum of neuroleptic-naive schizophrenic patients. Lancet. 1995;346:1130–1131. [PubMed] [Google Scholar]
19. Hietala J, Syvalahti E, Vilkman H, et al. Depressive symptoms and presynaptic dopamine function in neuroleptic-naive schizophrenia. Schizophr Res. 1999;35:41–50. [PubMed] [Google Scholar]
20. Howes OD, Montgomery AJ, Asselin MC, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2008 [PubMed] [Google Scholar]
21. Lindstrom LH, Gefvert O, Hagberg G, et al. Increased dopamine synthesis rate in medial prefrontal cortex and striatum in schizophrenia indicated by L-(beta-11C) DOPA and PET. Biol Psychiatry. 1999;46:681–688. [PubMed] [Google Scholar]
22. Reith J, Benkelfat C, Sherwin A, et al. Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci U S A. 1994;91:11651–11654. [PMC free article] [PubMed] [Google Scholar]
Reith J, Benkelfat C, Sherwin A, et al. Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci U S A. 1994;91:11651–11654. [PMC free article] [PubMed] [Google Scholar]
23. Howes OD, Montgomery AJ, Asselin MC, Murray RM, Grasby PM, McGuire PK. Molecular imaging studies of the striatal dopaminergic system in psychosis and predictions for the prodromal phase of psychosis. Br J Psychiatry Suppl. 2007;51:s13–s18. [PMC free article] [PubMed] [Google Scholar]
24. Dao-Castellana MH, Paillere-Martinot ML, Hantraye P, et al. Presynaptic dopaminergic function in the striatum of schizophrenic patients. Schizophr Res. 1997;23:167–174. [PubMed] [Google Scholar]
25. Elkashef AM, Doudet D, Bryant T, Cohen RM, Li SH, Wyatt RJ. 6-(18)F-DOPA PET study in patients with schizophrenia. Positron emission tomography. Psychiatry Res. 2000;100:1–11. [PubMed] [Google Scholar]
26. Laruelle M, Iyer RN, al-Tikriti MS, et al. Microdialysis and SPECT measurements of amphetamine-induced dopamine release in nonhuman primates. Synapse. 1997;25:1–14. [PubMed] [Google Scholar]
Synapse. 1997;25:1–14. [PubMed] [Google Scholar]
27. Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423–451. [PubMed] [Google Scholar]
28. Abi-Dargham A, Gil R, Krystal J, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155:761–767. [PubMed] [Google Scholar]
29. Breier A, Su TP, Saunders R, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A. 1997;94:2569–2574. [PMC free article] [PubMed] [Google Scholar]
30. Kestler LP, Walker E, Vega EM. Dopamine receptors in the brains of schizophrenia patients: a meta-analysis of the findings. Behav Pharmacol. 2001;12:355–371. [PubMed] [Google Scholar]
31. Laruelle M, Abi-Dargham A, van Dyck CH, et al. Single photon emission computerized tomography imaging of amphetamine-induced dopamine release in drug-free schizophrenic subjects.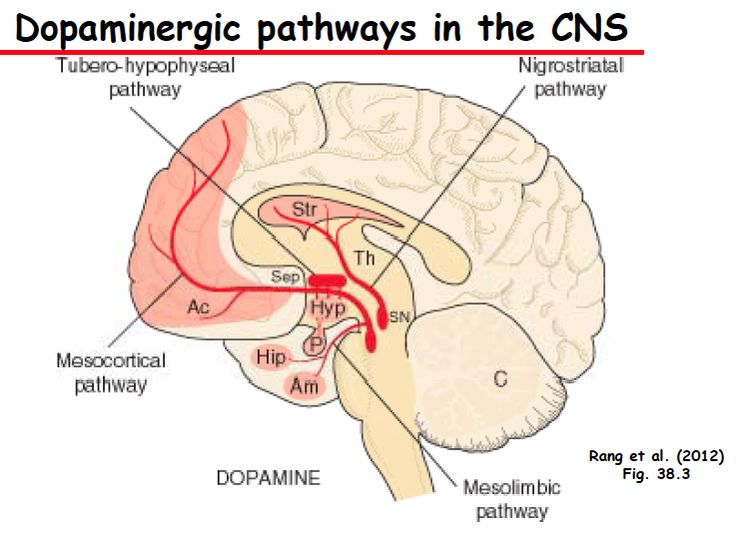 Proc Natl Acad Sci U S A. 1996;93:9235–9240. [PMC free article] [PubMed] [Google Scholar]
Proc Natl Acad Sci U S A. 1996;93:9235–9240. [PMC free article] [PubMed] [Google Scholar]
32. Laruelle M, Abi-Dargham A. Dopamine as the wind of the psychotic fire: new evidence from brain imaging studies. J Psychopharmacol. 1999;13:358–371. [PubMed] [Google Scholar]
33. Abi-Dargham A, Rodenhiser J, Printz D, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 2000;97:8104–8109. [PMC free article] [PubMed] [Google Scholar]
34. Crawley JC, Crow TJ, Johnstone EC, et al. Uptake of 77Br-spiperone in the striata of schizophrenic patients and controls. Nucl Med Commun. 1986;7:599–607. [PubMed] [Google Scholar]
35. Gjedde A, Wong DF. Positron tomographic quantitation of neuroreceptors in human brain in vivo–with special reference to the D2 dopamine receptors in caudate nucleus. Neurosurg Rev. 1987;10:9–18. [PubMed] [Google Scholar]
36. Wong DF, Wagner HN, Jr, Tune LE, et al. Positron emission tomography reveals elevated D2 dopamine receptors in drug-naive schizophrenics. Science. 1986 19;234:1558–1563. [PubMed] [Google Scholar]
Science. 1986 19;234:1558–1563. [PubMed] [Google Scholar]
37. Farde L, Wiesel FA, Stone-Elander S, et al. D2 dopamine receptors in neuroleptic-naive schizophrenic patients. A positron emission tomography study with [11C]raclopride. Arch Gen Psychiatry. 1990;47:213–219. [PubMed] [Google Scholar]
38. Martinot JL, Peron-Magnan P, Huret JD, et al. Striatal D2 dopaminergic receptors assessed with positron emission tomography and [76Br]bromospiperone in untreated schizophrenic patients. Am J Psychiatry. 1990;147:44–50. [PubMed] [Google Scholar]
39. Laruelle M. Imaging dopamine transmission in schizophrenia. A review and meta-analysis. Q J Nucl Med. 1998;42:211–221. [PubMed] [Google Scholar]
40. Zakzanis KK, Hansen KT. Dopamine D2 densities and the schizophrenic brain. Schizophr Res. 1998;32:201–206. [PubMed] [Google Scholar]
41. Okubo Y, Suhara T, Suzuki K, et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature. 1997;385:634–636. [PubMed] [Google Scholar]
[PubMed] [Google Scholar]
42. Karlsson P, Farde L, Halldin C, Sedvall G. PET study of D(1) dopamine receptor binding in neuroleptic-naive patients with schizophrenia. Am J Psychiatry. 2002;159:761–767. [PubMed] [Google Scholar]
43. Talvik M, Nordstrom AL, Okubo Y, et al. Dopamine D2 receptor binding in drug-naive patients with schizophrenia examined with raclopride-C11 and positron emission tomography. Psychiatry Res. 2006;148:165–173. [PubMed] [Google Scholar]
44. Buchsbaum MS, Christian BT, Lehrer DS, et al. D2/D3 dopamine receptor binding with [F-18]fallypride in thalamus and cortex of patients with schizophrenia. Schizophr Res. 2006;85:232–244. [PubMed] [Google Scholar]
45. Suhara T, Okubo Y, Yasuno F, et al. Decreased dopamine D2 receptor binding in the anterior cingulate cortex in schizophrenia. Arch Gen Psychiatry. 2002;59:25–30. [PubMed] [Google Scholar]
46. Takahashi H, Higuchi M, Suhara T. The role of extrastriatal dopamine D2 receptors in schizophrenia. Biol Psychiatry. 2006;59:919–928. [PubMed] [Google Scholar]
Biol Psychiatry. 2006;59:919–928. [PubMed] [Google Scholar]
47. Seeman P, Schwarz J, Chen JF, et al. Psychosis pathways converge via D2high dopamine receptors. Synapse. 2006;60:319–346. [PubMed] [Google Scholar]
48. Graf-Guerrero A, Romina M, Agid O, et al. The dopamine D2 receptors in high-affinity state and D3 receptors in schizophrenia: a clinical [11C]-(+)-PHNO PET study. Neuropsychopharmacology. 2009;34:1078–1086. [PubMed] [Google Scholar]
49. Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (Berl) 2004;174:3–16. [PubMed] [Google Scholar]
50. Tamminga CA. The neurobiology of cognition in schizophrenia. J Clin Psychiatry. 2006;67:e11. [PubMed] [Google Scholar]
51. Abi-Dargham A, Mawlawi O, Lombardo I, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–3719. [PMC free article] [PubMed] [Google Scholar]
52. Guo N, Hwang DR, Lo ES, Huang YY, Laruelle M, bi-Dargham A. Dopamine depletion and in vivo binding of PET D1 receptor radioligands: implications for imaging studies in schizophrenia. Neuropsychopharmacology. 2003;28:1703–1711. [PubMed] [Google Scholar]
Guo N, Hwang DR, Lo ES, Huang YY, Laruelle M, bi-Dargham A. Dopamine depletion and in vivo binding of PET D1 receptor radioligands: implications for imaging studies in schizophrenia. Neuropsychopharmacology. 2003;28:1703–1711. [PubMed] [Google Scholar]
53. Ekelund J, Slifstein M, Narendran R, et al. In vivo DA D(1) receptor selectivity of NNC 112 and SCH 23390. Mol Imaging Biol. 2007;9:117–125. [PubMed] [Google Scholar]
54. Frankle WG, Laruelle M. Neuroreceptor imaging in psychiatric disorders. Ann Nucl Med. 2002;16:437–446. [PubMed] [Google Scholar]
55. Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157:514–520. [PubMed] [Google Scholar]
56. Nordstrom AL, Farde L, Wiesel FA, et al. Central D2-dopamine receptor occupancy in relation to antipsychotic drug effects: a double-blind PET study of schizophrenic patients. Biol Psychiatry. 1993;33:227–235. [PubMed] [Google Scholar]
Biol Psychiatry. 1993;33:227–235. [PubMed] [Google Scholar]
57. Wolkin A, Barouche F, Wolf AP, et al. Dopamine blockade and clinical response: evidence for two biological subgroups of schizophrenia. Am J Psychiatry. 1989;146:905–908. [PubMed] [Google Scholar]
58. Grace AA, Bunney BS, Moore H, Todd CL. Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci. 1997;20:31–37. [PubMed] [Google Scholar]
59. Kapur S, Arenovich T, Agid O, Zipursky R, Lindborg S, Jones B. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am J Psychiatry. 2005;162:939–946. [PubMed] [Google Scholar]
60. Leucht S, Busch R, Hamann J, Kissling W, Kane JM. Early-onset hypothesis of antipsychotic drug action: a hypothesis tested, confirmed and extended. Biol Psychiatry. 2005;57:1543–1549. [PubMed] [Google Scholar]
61. Agid O, Mamo D, Ginovart N, et al. Striatal vs extrastriatal dopamine D2 receptors in antipsychotic response–a double-blind PET study in schizophrenia.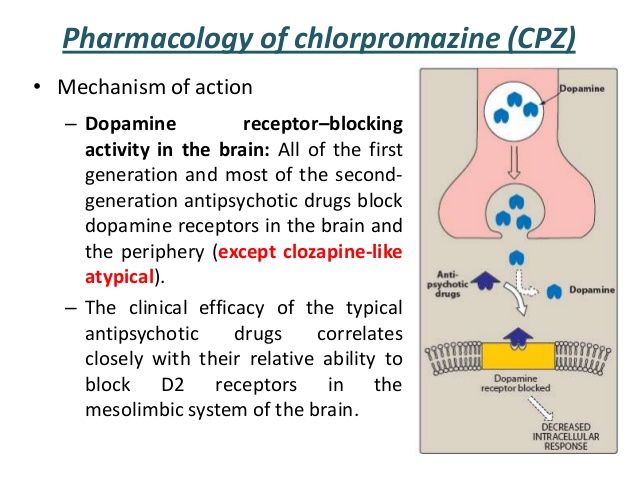 Neuropsychopharmacology. 2007;32:1209–1215. [PubMed] [Google Scholar]
Neuropsychopharmacology. 2007;32:1209–1215. [PubMed] [Google Scholar]
62. Catafau AM, Corripio I, Perez V, et al. Dopamine D2 receptor occupancy by risperidone: implications for the timing and magnitude of clinical response. Psychiatry Res. 2006;148:175–183. [PubMed] [Google Scholar]
63. Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. [PubMed] [Google Scholar]
64. Shi J, Gershon ES, Liu C. Genetic associations with schizophrenia: meta-analyses of 12 candidate genes. Schizophr Res. 2008;104:96–107. [PMC free article] [PubMed] [Google Scholar]
65. Stefansson H, Rujescu D, Cichon S, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. [PMC free article] [PubMed] [Google Scholar]
66. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. [PMC free article] [PubMed] [Google Scholar]
67. O'Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008 [PubMed] [Google Scholar]
O'Donovan MC, Craddock N, Norton N, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008 [PubMed] [Google Scholar]
68. Talkowski ME, Kirov G, Bamne M, et al. A network of dopaminergic gene variations implicated as risk factors for schizophrenia. Hum Mol Genet. 2008;17:747–758. [PMC free article] [PubMed] [Google Scholar]
69. Cantor-Graae E. The contribution of social factors to the development of schizophrenia: a review of recent findings. Can J Psychiatry. 2007;52:277–286. [PubMed] [Google Scholar]
70. van WR, Stefanis NC, Myin-Germeys I. Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene-stress interaction. Schizophr Bull. 2008 [PMC free article] [PubMed] [Google Scholar]
71. Hall FS, Wilkinson LS, Humby T, et al. Isolation rearing in rats: pre- and postsynaptic changes in striatal dopaminergic systems. Pharmacol Biochem Behav. 1998;59:859–872. [PubMed] [Google Scholar]
[PubMed] [Google Scholar]
72. Hall FS, Wilkinson LS, Humby T, Robbins TW. Maternal deprivation of neonatal rats produces enduring changes in dopamine function. Synapse. 1999;32:37–43. [PubMed] [Google Scholar]
73. Morgan D, Grant KA, Gage HD, et al. Social dominance in monkeys: dopamine D2 receptors and cocaine self-administration. Nat Neurosci. 2002;5:169–174. [PubMed] [Google Scholar]
74. Tidey JW, Miczek KA. Social defeat stress selectively alters mesocorticolimbic dopamine release: an in vivo microdialysis study. Brain Res. 1996;721:140–149. [PubMed] [Google Scholar]
75. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002;159:1080–1092. [PubMed] [Google Scholar]
76. Geddes JR, Lawrie SM. Obstetric complications and schizophrenia: a meta-analysis. Br J Psychiatry. 1995;167:786–793. [PubMed] [Google Scholar]
77. Kunugi H, Nanko S, Murray RM. Obstetric complications and schizophrenia: prenatal underdevelopment and subsequent neurodevelopmental impairment. Br J Psychiatry Suppl. 2001;40:s25–s29. [PubMed] [Google Scholar]
Br J Psychiatry Suppl. 2001;40:s25–s29. [PubMed] [Google Scholar]
78. Boksa P, El-Khodor BF. Birth insult interacts with stress at adulthood to alter dopaminergic function in animal models: possible implications for schizophrenia and other disorders. Neurosci Biobehav Rev. 2003;27:91–101. [PubMed] [Google Scholar]
79. Boksa P. Animal models of obstetric complications in relation to schizophrenia. Brain Res Brain Res Rev. 2004;45:1–17. [PubMed] [Google Scholar]
80. Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. [PubMed] [Google Scholar]
81. Lipska BK, Halim ND, Segal PN, Weinberger DR. Effects of reversible inactivation of the neonatal ventral hippocampus on behavior in the adult rat. J Neurosci. 2002;22:2835–2842. [PMC free article] [PubMed] [Google Scholar]
82. Flores G, Wood GK, Liang JJ, Quirion R, Srivastava LK.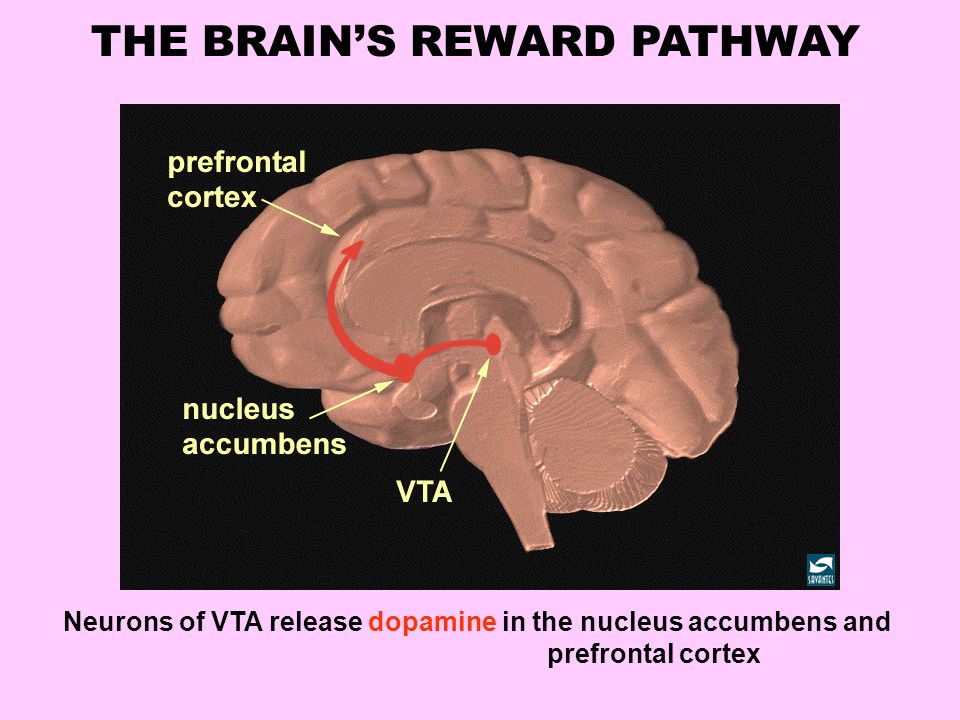 Enhanced amphetamine sensitivity and increased expression of dopamine D2 receptors in postpubertal rats after neonatal excitotoxic lesions of the medial prefrontal cortex. J Neurosci. 1996;16:7366–7375. [PMC free article] [PubMed] [Google Scholar]
Enhanced amphetamine sensitivity and increased expression of dopamine D2 receptors in postpubertal rats after neonatal excitotoxic lesions of the medial prefrontal cortex. J Neurosci. 1996;16:7366–7375. [PMC free article] [PubMed] [Google Scholar]
83. Diaz R, Ogren SO, Blum M, Fuxe K. Prenatal corticosterone increases spontaneous and d-amphetamine induced locomotor activity and brain dopamine metabolism in prepubertal male and female rats. Neuroscience. 1995;66:467–473. [PubMed] [Google Scholar]
84. Henry C, Guegant G, Cador M, et al. Prenatal stress in rats facilitates amphetamine-induced sensitization and induces long-lasting changes in dopamine receptors in the nucleus accumbens. Brain Res. 1995;685:179–186. [PubMed] [Google Scholar]
85. Fortier ME, Joober R, Luheshi GN, Boksa P. Maternal exposure to bacterial endotoxin during pregnancy enhances amphetamine-induced locomotion and startle responses in adult rat offspring. J Psychiatr Res. 2004;38:335–345. [PubMed] [Google Scholar]
86. Watanabe M, Nonaka R, Hagino Y, Kodama Y. Effects of prenatal methylazoxymethanol treatment on striatal dopaminergic systems in rat brain. Neurosci Res. 1998;30:135–144. [PubMed] [Google Scholar]
Watanabe M, Nonaka R, Hagino Y, Kodama Y. Effects of prenatal methylazoxymethanol treatment on striatal dopaminergic systems in rat brain. Neurosci Res. 1998;30:135–144. [PubMed] [Google Scholar]
87. Kehoe P, Shoemaker WJ, Triano L, Hoffman J, Arons C. Repeated isolation in the neonatal rat produces alterations in behavior and ventral striatal dopamine release in the juvenile after amphetamine challenge. Behav Neurosci. 1996;110:1435–1444. [PubMed] [Google Scholar]
88. Kehoe P, Clash K, Skipsey K, Shoemaker WJ. Brain dopamine response in isolated 10-day-old rats: assessment using D2 binding and dopamine turnover. Pharmacol Biochem Behav. 1996;53:41–49. [PubMed] [Google Scholar]
89. Angrist BM, Gershon S. The phenomenology of experimentally induced amphetamine psychosis–preliminary observations. Biol Psychiatry. 1970;2:95–107. [PubMed] [Google Scholar]
90. Yui K, Ikemoto S, Ishiguro T, Goto K. Studies of amphetamine or methamphetamine psychosis in Japan: relation of methamphetamine psychosis to schizophrenia.![]() Ann N Y Acad Sci. 2000;914:1–12. [PubMed] [Google Scholar]
Ann N Y Acad Sci. 2000;914:1–12. [PubMed] [Google Scholar]
91. Boileau I, Dagher A, Leyton M, et al. Modeling sensitization to stimulants in humans: an [11C]raclopride/positron emission tomography study in healthy men. Arch Gen Psychiatry. 2006;63:1386–1395. [PubMed] [Google Scholar]
92. Arseneault L, Cannon M, Witton J, Murray RM. Causal association between cannabis and psychosis: examination of the evidence. Br J Psychiatry. 2004;184:110–117. [PubMed] [Google Scholar]
93. Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370:319–328. [PubMed] [Google Scholar]
94. Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. [PubMed] [Google Scholar]
95. Cheer JF, Wassum KM, Heien ML, Phillips PE, Wightman RM. Cannabinoids enhance subsecond dopamine release in the nucleus accumbens of awake rats. J Neurosci. 2004;24:4393–4400. [PMC free article] [PubMed] [Google Scholar]
2004;24:4393–4400. [PMC free article] [PubMed] [Google Scholar]
96. Tanda G, Pontieri FE, Di CG. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science. 1997;276:2048–2050. [PubMed] [Google Scholar]
97. Bossong MG, van Berckel BN, Boellaard R, et al. Delta9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology. 2008 [PubMed] [Google Scholar]
98. Bowers MB, Kantrowitz JT. Elevated plasma dopamine metabolites in cannabis psychosis. Am J Psychiatry. 2007;164:1615–1616. [PubMed] [Google Scholar]
99. Kegeles LS, Abi-Dargham A, Zea-Ponce Y, et al. Modulation of amphetamine-induced striatal dopamine release by ketamine in humans: implications for schizophrenia. Biol Psychiatry. 2000;48:627–640. [PubMed] [Google Scholar]
100. Cannon TD, van Erp TG, Rosso IM, et al. Fetal hypoxia and structural brain abnormalities in schizophrenic patients, their siblings, and controls. Arch Gen Psychiatry. 2002;59:35–41. [PubMed] [Google Scholar]
Arch Gen Psychiatry. 2002;59:35–41. [PubMed] [Google Scholar]
101. Nicodemus KK, Marenco S, Batten AJ, et al. Serious obstetric complications interact with hypoxia-regulated/vascular-expression genes to influence schizophrenia risk. Mol Psychiatry. 2008;13:873–877. [PubMed] [Google Scholar]
102. Mittal VA, Ellman LM, Cannon TD. Gene-environment interaction and covariation in schizophrenia: the role of obstetric complications. Schizophr Bull. 2008 [PMC free article] [PubMed] [Google Scholar]
103. van Os J, Rutten BP, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34:1066–1082. [PMC free article] [PubMed] [Google Scholar]
104. Howes SR, Dalley JW, Morrison CH, Robbins TW, Everitt BJ. Leftward shift in the acquisition of cocaine self-administration in isolation-reared rats: relationship to extracellular levels of dopamine, serotonin and glutamate in the nucleus accumbens and amygdala-striatal FOS expression. Psychopharmacology (Berl) 2000;151:55–63. [PubMed] [Google Scholar]
Psychopharmacology (Berl) 2000;151:55–63. [PubMed] [Google Scholar]
105. Jones GH. Social isolation and individual differences: behavioural and dopaminergic responses to psychomotor stimulants. Clin Neuropharmacol. 1992;15(suppl 1, pt A):253A–254A. [PubMed] [Google Scholar]
106. Fulford AJ, Marsden CA. Effect of isolation-rearing on conditioned dopamine release in vivo in the nucleus accumbens of the rat. J Neurochem. 1998;70:384–390. [PubMed] [Google Scholar]
107. Pruessner JC, Champagne F, Meaney MJ, Dagher A. Dopamine release in response to a psychological stress in humans and its relationship to early life maternal care: a positron emission tomography study using [11C]raclopride. J Neurosci. 2004;24:2825–2831. [PMC free article] [PubMed] [Google Scholar]
108. Kegeles LS, Martinez D, Kochan LD, et al. NMDA antagonist effects on striatal dopamine release: positron emission tomography studies in humans. Synapse. 2002;43:19–29. [PubMed] [Google Scholar]
109. Wassef A, Baker J, Kochan LD. GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol. 2003;23:601–640. [PubMed] [Google Scholar]
Wassef A, Baker J, Kochan LD. GABA and schizophrenia: a review of basic science and clinical studies. J Clin Psychopharmacol. 2003;23:601–640. [PubMed] [Google Scholar]
110. Caspi A, Moffitt TE, Cannon M, et al. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry. 2005;57:1117–1127. [PubMed] [Google Scholar]
111. van Os J, Pedersen CB, Mortensen PB. Confirmation of synergy between urbanicity and familial liability in the causation of psychosis. Am J Psychiatry. 2004;161:2312–2314. [PubMed] [Google Scholar]
112. van Os J, Hanssen M, Bak M, Bijl RV, Vollebergh W. Do urbanicity and familial liability coparticipate in causing psychosis? Am J Psychiatry. 2003;160:477–482. [PubMed] [Google Scholar]
113. Cantor-Graae E, Selten JP. Schizophrenia and migration: a meta-analysis and review. Am J Psychiatry.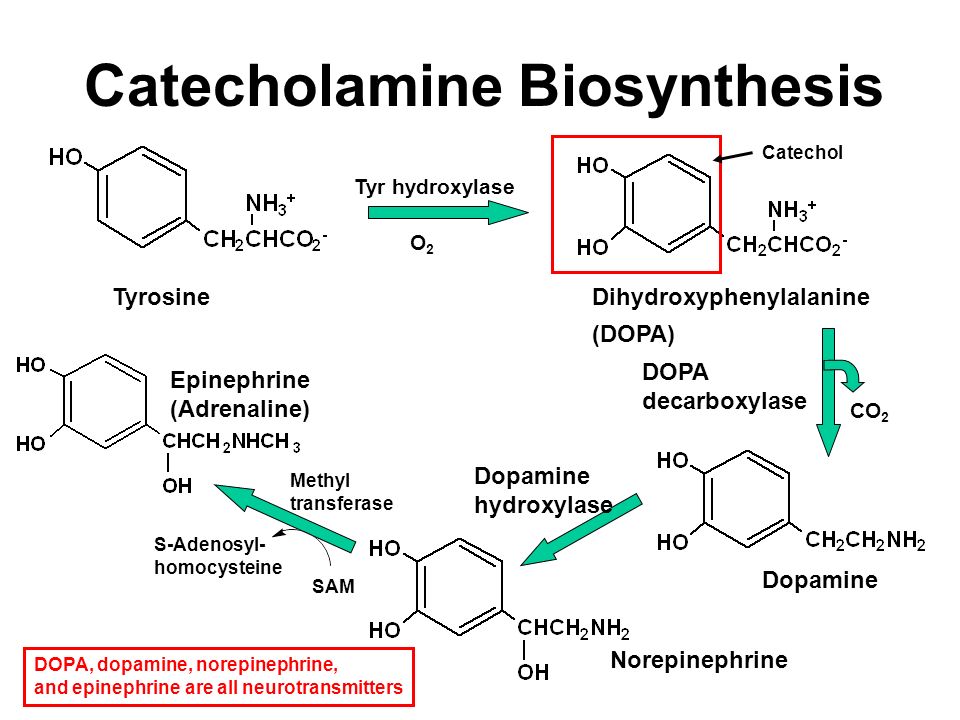 2005;162:12–24. [PubMed] [Google Scholar]
2005;162:12–24. [PubMed] [Google Scholar]
114. McKetin R, McLaren J, Lubman DI, Hides L. The prevalence of psychotic symptoms among methamphetamine users. Addiction. 2006;101:1473–1478. [PubMed] [Google Scholar]
115. Cannon TD, Cadenhead K, Cornblatt B, et al. Prediction of psychosis in youth at high clinical risk: a multisite longitudinal study in North America. Arch Gen Psychiatry. 2008;65:28–37. [PMC free article] [PubMed] [Google Scholar]
116. Yung AR. Psychosis prediction: 12-month follow up of a high-risk (“prodromal”) group. 2003 [PubMed] [Google Scholar]
117. Abi-Dargham A, Kegeles LS, Zea-Ponce Y, et al. Striatal amphetamine-induced dopamine release in patients with schizotypal personality disorder studied with single photon emission computed tomography and [123I]iodobenzamide. Biol Psychiatry. 2004;55:1001–1006. [PubMed] [Google Scholar]
118. Soliman A, O'Driscoll GA, Pruessner J, et al. Stress-induced dopamine release in humans at risk of psychosis: a [(11)C]raclopride PET study.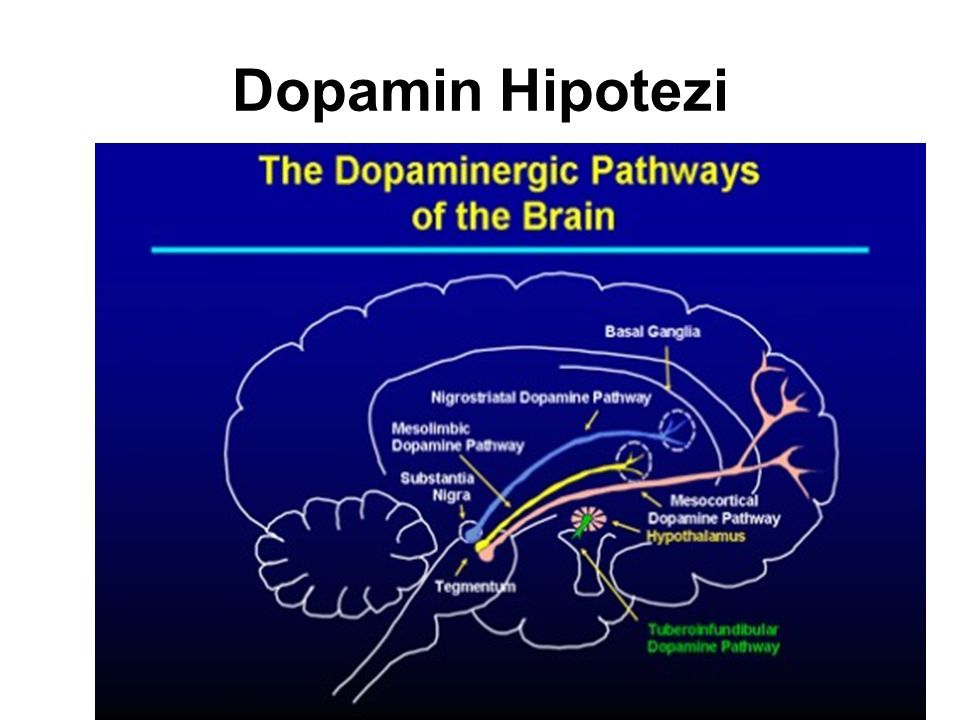 Neuropsychopharmacology. 2008;33:2033–2041. [PubMed] [Google Scholar]
Neuropsychopharmacology. 2008;33:2033–2041. [PubMed] [Google Scholar]
119. Huttunen J, Heinimaa M, Svirskis T, et al. Striatal dopamine synthesis in first-degree relatives of patients with schizophrenia. Biol Psychiatry. 2007 [PubMed] [Google Scholar]
120. Brunelin J, d'Amato T, van Os J, Cochet A, Suaud-Chagny MF, Saoud M. Effects of acute metabolic stress on the dopaminergic and pituitary-adrenal axis activity in patients with schizophrenia, their unaffected siblings and controls. Schizophr Res. 2008;100:206–211. [PubMed] [Google Scholar]
121. Myin-Germeys I, Marcelis M, Krabbendam L, Delespaul P, van Os J. Subtle fluctuations in psychotic phenomena as functional states of abnormal dopamine reactivity in individuals at risk. Biol Psychiatry. 2005;58:105–110. [PubMed] [Google Scholar]
122. Wood SJ, Pantelis C, Velakoulis D, Yucel M, Fornito A, McGorry PD. Progressive changes in the development toward schizophrenia: studies in subjects at increased symptomatic risk. Schizophr Bull. 2008;34:322–329. [PMC free article] [PubMed] [Google Scholar]
Schizophr Bull. 2008;34:322–329. [PMC free article] [PubMed] [Google Scholar]
123. Boos HB, Aleman A, Cahn W, Pol HH, Kahn RS. Brain volumes in relatives of patients with schizophrenia: a meta-analysis. Arch Gen Psychiatry. 2007;64:297–304. [PubMed] [Google Scholar]
124. Dickey CC, McCarley RW, Shenton ME. The brain in schizotypal personality disorder: a review of structural MRI and CT findings. Harv Rev Psychiatry. 2002;10:1–15. [PMC free article] [PubMed] [Google Scholar]
125. Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. [PMC free article] [PubMed] [Google Scholar]
126. DeLisi LE. The concept of progressive brain change in schizophrenia: implications for understanding schizophrenia. Schizophr Bull. 2008;34:312–321. [PMC free article] [PubMed] [Google Scholar]
127. van Haren NE, Hulshoff Pol HE, Schnack HG, et al. Focal gray matter changes in schizophrenia across the course of the illness: a 5-year follow-up study. Neuropsychopharmacology. 2007;32:2057–2066. [PubMed] [Google Scholar]
Neuropsychopharmacology. 2007;32:2057–2066. [PubMed] [Google Scholar]
128. Brans RG, van Haren NE, van Baal GC, Schnack HG, Kahn RS, Hulshoff Pol HE. Heritability of changes in brain volume over time in twin pairs discordant for schizophrenia. Arch Gen Psychiatry. 2008;65:1259–1268. [PubMed] [Google Scholar]
129. Lieberman JA, Tollefson GD, Charles C, et al. Antipsychotic drug effects on brain morphology in first-episode psychosis. Arch Gen Psychiatry. 2005;62:361–370. [PubMed] [Google Scholar]
130. Konopaske GT, Dorph-Petersen KA, Pierri JN, Wu Q, Sampson AR, Lewis DA. Effect of chronic exposure to antipsychotic medication on cell numbers in the parietal cortex of macaque monkeys. Neuropsychopharmacology. 2007;32:1216–1223. [PubMed] [Google Scholar]
131. Rais M, Cahn W, Van HN, et al. Excessive brain volume loss over time in cannabis-using first-episode schizophrenia patients. Am J Psychiatry. 2008;165:490–496. [PubMed] [Google Scholar]
132. Fusar-Poli P, Perez J, Broome M, et al. Neurofunctional correlates of vulnerability to psychosis: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2007;31:465–484. [PubMed] [Google Scholar]
Neurofunctional correlates of vulnerability to psychosis: a systematic review and meta-analysis. Neurosci Biobehav Rev. 2007;31:465–484. [PubMed] [Google Scholar]
133. Lawrie SM, McIntosh AM, Hall J, Owens DG, Johnstone EC. Brain structure and function changes during the development of schizophrenia: the evidence from studies of subjects at increased genetic risk. Schizophr Bull. 2008;34:330–340. [PMC free article] [PubMed] [Google Scholar]
134. Brewer WJ, Wood SJ, Phillips LJ, et al. Generalized and specific cognitive performance in clinical high-risk cohorts: a review highlighting potential vulnerability markers for psychosis. Schizophr Bull. 2006;32:538–555. [PMC free article] [PubMed] [Google Scholar]
135. Eastvold AD, Heaton RK, Cadenhead KS. Neurocognitive deficits in the (putative) prodrome and first episode of psychosis. Schizophr Res. 2007;93:266–277. [PMC free article] [PubMed] [Google Scholar]
136. Simon AE, Cattapan-Ludewig K, Zmilacher S, et al. Cognitive functioning in the schizophrenia prodrome.![]() Schizophr Bull. 2007;33:761–771. [PMC free article] [PubMed] [Google Scholar]
Schizophr Bull. 2007;33:761–771. [PMC free article] [PubMed] [Google Scholar]
137. Laruelle M. Schizophrenia is associated with increased synaptic dopamine in associative rather than limbic regions of the striatum: implications for the mechanisms of actions of antipsychotic drugs. Schizophr Res. 2006;81:16. [Google Scholar]
138. Dutta R, Greene T, Addington J, McKenzie K, Phillips M, Murray RM. Biological, life course, and cross-cultural studies all point toward the value of dimensional and developmental ratings in the classification of psychosis. Schizophr Bull. 2007;33:868–876. [PMC free article] [PubMed] [Google Scholar]
139. Peralta V, Cuesta MJ. How many and which are the psychopathological dimensions in schizophrenia? Issues influencing their ascertainment. Schizophr Res. 2001;49:269–285. [PubMed] [Google Scholar]
140. Kessler RC, Birnbaum H, Demler O, et al. The prevalence and correlates of nonaffective psychosis in the National Comorbidity Survey Replication (NCS-R) Biol Psychiatry. 2005;58:668–676. [PMC free article] [PubMed] [Google Scholar]
2005;58:668–676. [PMC free article] [PubMed] [Google Scholar]
141. van Os J, Linscott RJ, Myin-Germeys I, Delespaul P, Krabbendam L. A systematic review and meta-analysis of the psychosis continuum: evidence for a psychosis proneness-persistence-impairment model of psychotic disorder. Psychol Med. 2008:1–17. [PubMed] [Google Scholar]
142. Yatham LN, Liddle PF, Shiah IS, et al. PET study of [(18)F]6-fluoro-L-dopa uptake in neuroleptic- and mood-stabilizer-naive first-episode nonpsychotic mania: effects of treatment with divalproex sodium. Am J Psychiatry. 2002;159:768–774. [PubMed] [Google Scholar]
143. Martinot M, Bragulat V, Artiges E, et al. Decreased presynaptic dopamine function in the left caudate of depressed patients with affective flattening and psychomotor retardation. Am J Psychiatry. 2001;158:314–316. [PubMed] [Google Scholar]
144. Parsey RV, Oquendo MA, Zea-Ponce Y, et al. Dopamine D(2) receptor availability and amphetamine-induced dopamine release in unipolar depression.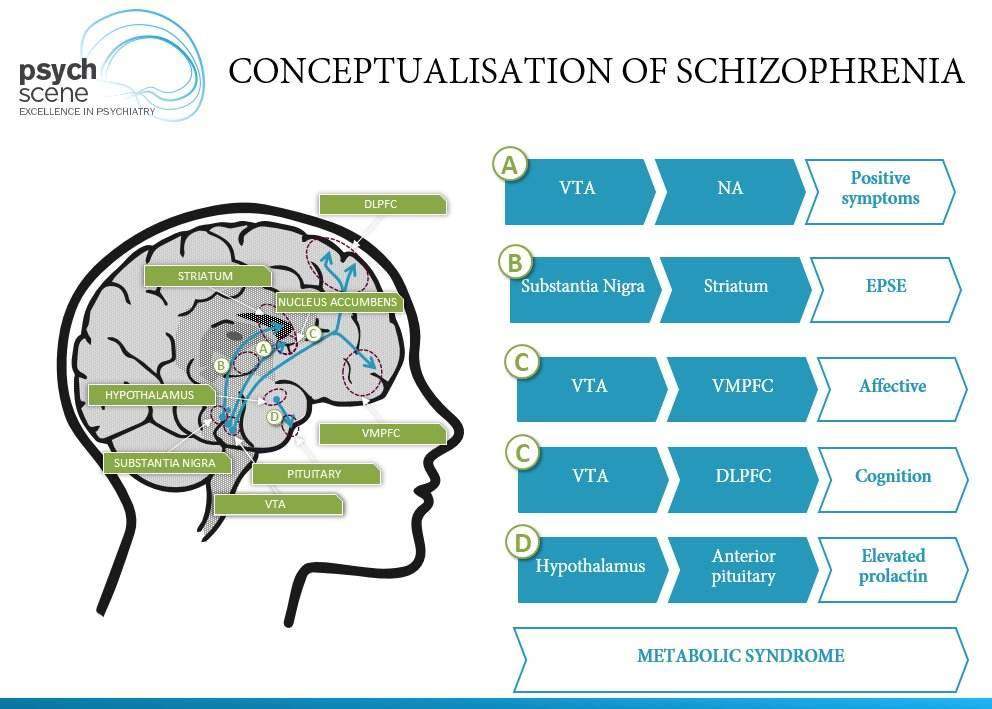 Biol Psychiatry. 2001;50:313–322. [PubMed] [Google Scholar]
Biol Psychiatry. 2001;50:313–322. [PubMed] [Google Scholar]
145. Reith J, Benkelfat C, Sherwin A, et al. Elevated dopa decarboxylase activity in living brain of patients with psychosis. Proc Natl Acad Sci U S A. 1994;91:11651–11654. [PMC free article] [PubMed] [Google Scholar]
146. Turjanski N, Sawle GV, Playford ED, et al. PET studies of the presynaptic and postsynaptic dopaminergic system in Tourette's syndrome. J Neurol Neurosurg Psychiatry. 1994;57:688–692. [PMC free article] [PubMed] [Google Scholar]
147. Ernst M, Zametkin AJ, Matochik JA, Pascualvaca D, Cohen RM. Low medial prefrontal dopaminergic activity in autistic children. Lancet. 1997;350:638. [PubMed] [Google Scholar]
148. Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. [PubMed] [Google Scholar]
149. Dannon PN, Lowengrub K, Gonopolski Y, Kotler M. Current and emerging somatic treatment strategies in psychotic major depression. Expert Rev Neurother. 2006;6:73–80. [PubMed] [Google Scholar]
Expert Rev Neurother. 2006;6:73–80. [PubMed] [Google Scholar]
150. Zahodne LB, Fernandez HH. Pathophysiology and treatment of psychosis in Parkinson's disease: a review. Drugs Aging. 2008;25:665–682. [PMC free article] [PubMed] [Google Scholar]
151. Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–69. [PubMed] [Google Scholar]
152. Robbins TW, Everitt BJ. Functional studies of the central catecholamines. Int Rev Neurobiol. 1982;23:303–365. [PubMed] [Google Scholar]
153. Robbins TW, Everitt BJ. Neurobehavioural mechanisms of reward and motivation. Curr Opin Neurobiol. 1996;6:228–236. [PubMed] [Google Scholar]
154. Heinz A. Anhedonia–a general nosology surmounting correlate of a dysfunctional dopaminergic reward system? Nervenarzt. 1999;70:391–398. [PubMed] [Google Scholar]
155. Martin-Soelch C, Leenders KL, Chevalley AF, et al. Reward mechanisms in the brain and their role in dependence: evidence from neurophysiological and neuroimaging studies.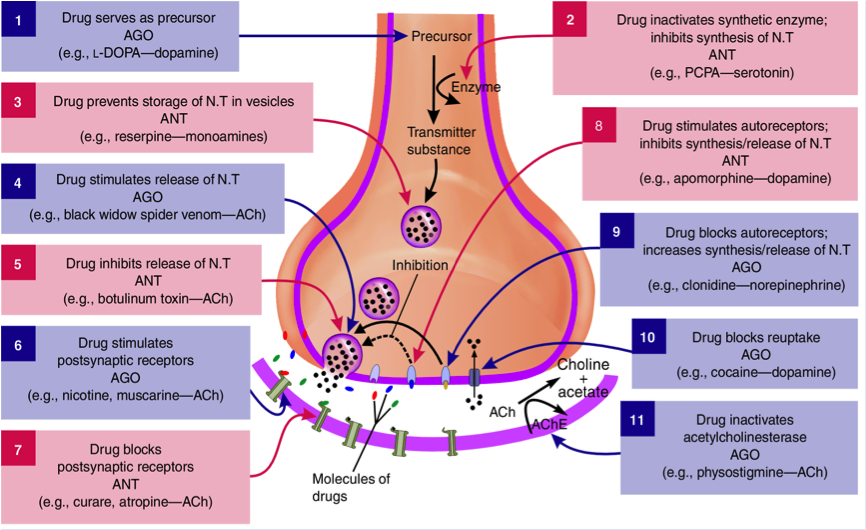 Brain Res Brain Res Rev. 2001;36:139–149. [PubMed] [Google Scholar]
Brain Res Brain Res Rev. 2001;36:139–149. [PubMed] [Google Scholar]
156. Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. [PubMed] [Google Scholar]
157. Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. [PubMed] [Google Scholar]
158. Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. [PubMed] [Google Scholar]
159. Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. [PubMed] [Google Scholar]
160. Kapur S, Mizrahi R, Li M. From dopamine to salience to psychosis–linking biology, pharmacology and phenomenology of psychosis. Schizophr Res. 2005;79:59–68. [PubMed] [Google Scholar]
161. Roiser JP, Stephan KE, Ouden HE, Barnes TR, Friston KJ, Joyce EM. Do patients with schizophrenia exhibit aberrant salience? Psychol Med. 2008:1–11. [PMC free article] [PubMed] [Google Scholar]
162. Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. [PubMed] [Google Scholar]
Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. [PubMed] [Google Scholar]
163. Juckel G, Schlagenhauf F, Koslowski M, et al. Dysfunction of ventral striatal reward prediction in schizophrenia. Neuroimage. 2006;29:409–416. [PubMed] [Google Scholar]
164. Cannon TD, van Erp TG, Bearden CE, et al. Early and late neurodevelopmental influences in the prodrome to schizophrenia: contributions of genes, environment, and their interactions. Schizophr Bull. 2003;29:653–669. [PubMed] [Google Scholar]
165. Howes OD, McDonald C, Cannon M, Arseneault L, Boydell J, Murray RM. Pathways to schizophrenia: the impact of environmental factors. Int J Neuropsychopharmacol. 2004;7(suppl 1):S7–S13. [PubMed] [Google Scholar]
166. Vernaleken I, Kumakura Y, Cumming P, et al. Modulation of [18F]fluorodopa (FDOPA) kinetics in the brain of healthy volunteers after acute haloperidol challenge. Neuroimage.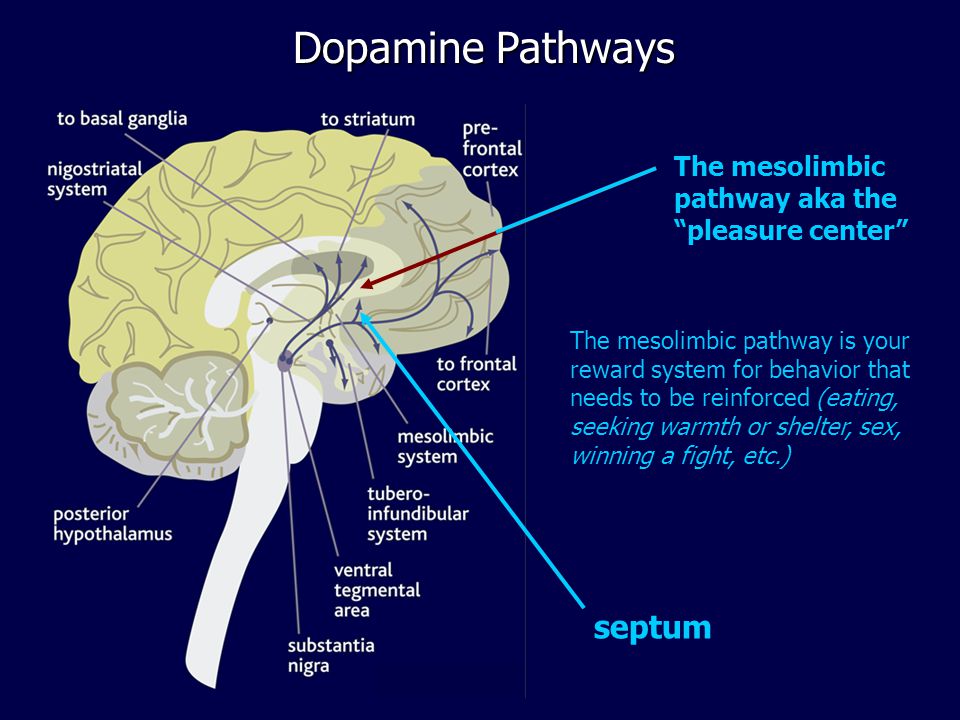 2006;30:1332–1339. [PubMed] [Google Scholar]
2006;30:1332–1339. [PubMed] [Google Scholar]
167. Grunder G, Vernaleken I, Muller MJ, et al. Subchronic haloperidol downregulates dopamine synthesis capacity in the brain of schizophrenic patients in vivo. Neuropsychopharmacology. 2003;28:787–794. [PubMed] [Google Scholar]
168. Meyer-Lindenberg A, Miletich RS, Kohn PD, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nat Neurosci. 2002;5:267–271. [PubMed] [Google Scholar]
169. Fusar-Poli P, Howes OD, Allen P, et al. Altered prefrontal activation directly related to striatal dopamine dysfunction in people with prodromal symptoms of schizophrenia. 2008;102 S30. [Google Scholar]
170. Kellendonk C, Simpson EH, Polan HJ, et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron. 2006;49:603–615. [PubMed] [Google Scholar]
171. Patil ST, Zhang L, Martenyi F, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. [PubMed] [Google Scholar]
Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. [PubMed] [Google Scholar]
The Dopamine Hypothesis of Schizophrenia
The dopamine hypothesis stems from early research carried out in the 1960’s and 1970’s when studies involved the use of amphetamine (increases dopamine levels) which increased psychotic symptoms while reserpine which depletes dopamine levels reduced psychotic symptoms.
The original dopamine hypothesis was put forward by Van Rossum in 1967 that stated that there was hyperactivity of dopamine transmission, which resulted in symptoms of schizophrenia and drugs that blocked dopamine reduced psychotic symptoms. [1]
DOPAMINE PRODUCTION AND METABOLISM
Dopamine is synthesised from the amino acid tyrosine. Tyrosine is converted into DOPA by the enzyme tyrosine hydroxylase.
DOPA is converted into dopamine (DA) by the enzyme DOPA decarboxylase (DOPADC).
This dopamine is packed and stored into synaptic vesicles via the vesicular monoamine transporter (VMAT2) and stored until its release into the synapse.
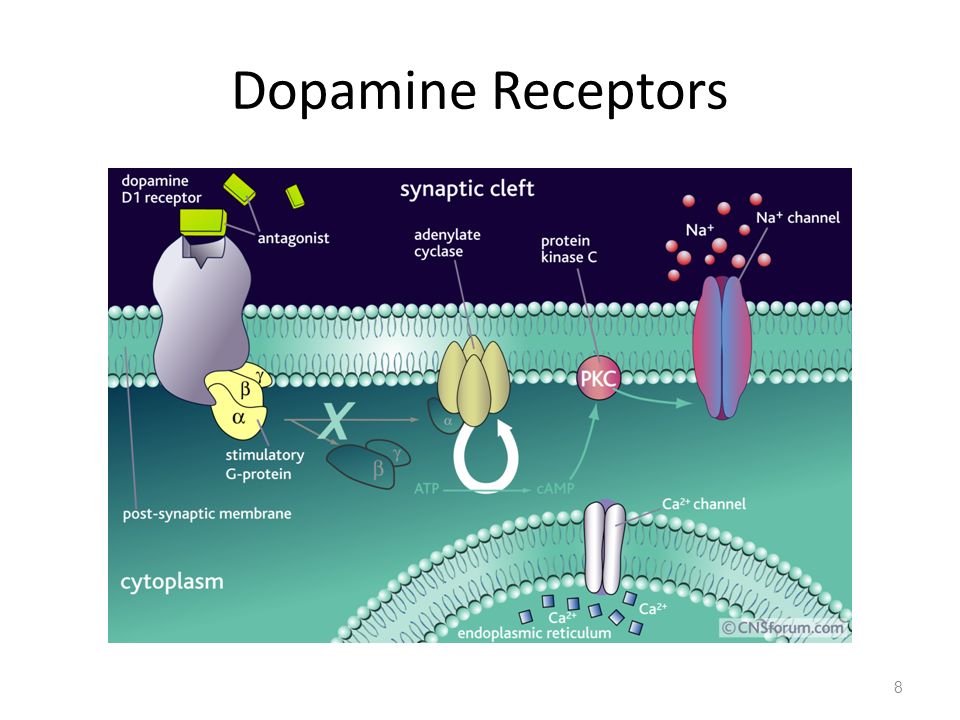
Dopamine Receptors:
When dopamine is released during neurotransmission, it acts on 5 types of postsynaptic receptors (D1-D5).
A negative feedback mechanism exists through the presynaptic D2 receptor which regulates the release of dopamine from the presynaptic neuron.
Dopamine Breakdown
Any excess dopamine is also ‘mopped up’ from the synapse by Dopamine transporter (DAT) and stored in the vesicles via VMAT2.
Dopamine is broken down by monoamine oxidase A (MAO-A), MAO-B and catechol-o-methyltransferase (COMT).
Learning points:
- Tyrosine hydroxylase is the rate-limiting step in the production of dopamine. Its expression is significantly increased in the substantia nigra of schizophrenia patients when compared to normal healthy subjects.
 [2]
[2] - Carbidopa is a peripheral DOPA-decarboxylase inhibitor co-administered with levodopa. Carbidopa prevents the conversion of levodopa to dopamine in the periphery, thus allowing more levodopa to pass the blood-brain barrier to be converted into dopamine for its therapeutic effect.
- Methamphetamine increases extracellular dopamine by interacting at vesicular monoamine transporter-2 (VMAT2) to inhibit dopamine uptake and promote dopamine release from synaptic vesicles, increasing cytosolic dopamine available for reverse transport by the dopamine transporter (DAT).
- Valbenazine a highly selective VMAT2 inhibitor has been approved by the FDA for the treatment of tardive dyskinesia.
- There is compelling evidence that presynaptic dopamine dysfunction results in increased availability and release of dopamine and this has been shown to be associated with prodromal symptoms of schizophrenia. Furthermore, dopamine synthesis capacity has also been shown to steadily increase with the onset of severe psychotic symptoms.
 [3] , [Howes & Shatalina, 2022]
[3] , [Howes & Shatalina, 2022]
- Dopaminergic transmission in the prefrontal cortex is mainly mediated by D1 receptors, and D1 dysfunction has been linked to cognitive impairment and negative symptoms of schizophrenia. [4]
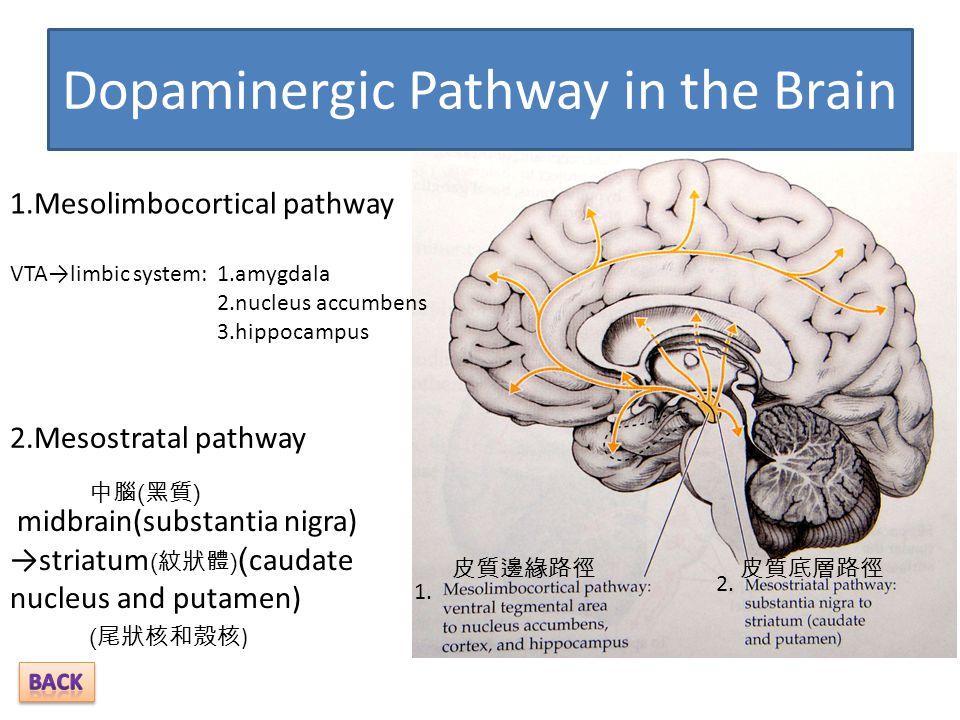
THE 4 DOPAMINE PATHWAYS IN THE BRAIN
1.The Mesolimbic Pathway
- The pathway projects from the ventral tegmental area (VTA) to the nucleus accumbens in the limbic system.
- Hyperactivity of dopamine in the mesolimbic pathway mediates positive psychotic symptoms. The pathway may also mediate aggression.
- The mesolimbic pathway is also the site of the rewards pathway and mediates pleasure and reward. Antipsychotics can block D2 receptors in this pathway reducing pleasure effects. This may be one explanation as to why individuals with schizophrenia have a higher incidence of smoking as nicotine enhances dopamine in the reward pathway (self-medication hypothesis)
- Antagonism of D2 receptors in the mesolimbic pathway treats positive psychotic symptoms.

- There is an occupancy requirement with the minimum threshold at 65% occupancy for treatment to be effective. Observations support this relationship between D2-receptor occupancy and clinical response that 80% of responders have D2-receptor occupancy above this threshold after treatment. [5]
2.The Mesocortical Pathway
- Projects from the VTA to the prefrontal cortex.
- Projections to the dorsolateral prefrontal cortex regulate cognition and executive functioning.
- Projections into the ventromedial prefrontal cortex regulate emotions and affect.
- Decreased dopamine in the mesocortical projection to the dorsolateral prefrontal cortex is postulated to be responsible for negative and depressive symptoms of schizophrenia.
- Nicotine releases dopamine in the mesocortical pathways alleviating negative symptoms (self-medication hypothesis).
3.The Nigrostriatal Pathway
- Projects from the dopaminergic neurons in the substantia nigra to the basal ganglia or striatum.

- The nigrostriatal pathway mediates motor movements.
- Blockade of dopamine D2 receptors in this pathway can lead to dystonia, parkinsonian symptoms and akathisia.
- Hyperactivity of dopamine in the nigrostriatal pathway is the postulated mechanism in hyperkinetic movement disorders such as chorea, tics and dyskinesias.
- Long-standing D2 blockade in the nigrostriatal pathway can lead to tardive dyskinesia.
4.The Tuberoinfundibular (TI) Pathway
- Projects from the hypothalamus to the anterior pituitary.
- The TI pathway inhibits prolactin release.
- Blockade of D2 receptors in this pathway can lead to hyperprolactinemia which clinically manifests as amenorrhoea, galactorrhoea and sexual dysfunction.
- Long-term hyperprolactinemia can be associated with osteoporosis.
Conceptualisation of Schizophrenia
Based on the above understanding, schizophrenia is best conceptualised as a complex entity which involves multiple pathways.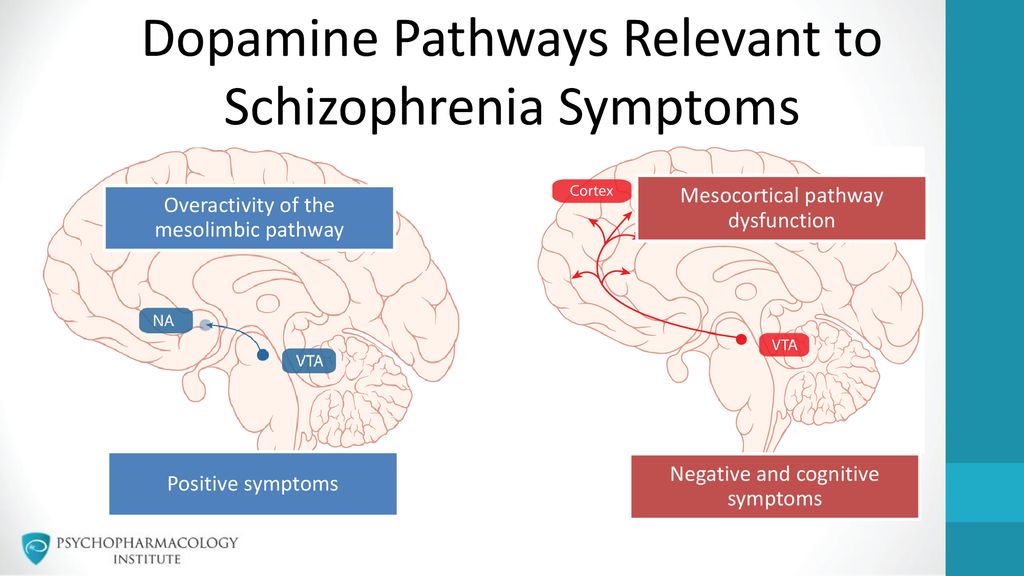
In clinical practice, there can be a disproportionate focus on positive psychotic symptoms.
It is however, important to recognise that affective (e.g depressive), negative and cognitive symptoms are a core part of schizophrenia and should be taken into account in treatment.
The aim of treatment, thus, is to modulate treatment creating a balance between effectiveness and reduction of side effects.
The balance is achieved by optimal dopamine blockade in the mesolimbic pathway while preserving (or enhancing) dopamine transmission in the other pathways.
DOPAMINE AND SCHIZOPHRENIA
The dopamine hypothesis of schizophrenia has moved from the dopamine receptor hypothesis (increased dopamine transmission at the postsynaptic receptors) to a focus on presynaptic striatal hyperdopaminergia.
According to Howes and Kapur-
This hypothesis accounts for the multiple environmental and genetic risk factors for schizophrenia and proposes that these interact to funnel through one final common pathway of presynaptic striatal hyperdopaminergia.

In addition to funneling through dopamine dysregulation, the multiple environmental and genetic risk factors influence diagnosis by affecting other aspects of brain function that underlie negative and cognitive symptoms. Schizophrenia is thus dopamine dysregulation in the context of a compromised brain. [6]
Read more on the molecular imaging of dopamine abnormalities in schizophrenia.
Clinical Implications
The hypothesis that the final common pathway is presynaptic dopamine dysregulation has some important clinical implications. Firstly, it implies that current antipsychotic drugs are not treating the primary abnormality and are acting downstream. While antipsychotic drugs block the effect of inappropriate dopamine release, they may paradoxically worsen the primary abnormality by blocking presynaptic D2 autoreceptors, resulting in a compensatory increase in dopamine synthesis.
This may explain why patients relapse rapidly on stopping their medication, and if the drugs may even worsen the primary abnormality, it also accounts for more severe relapse after discontinuing treatment.
This suggests that drug development needs to focus on modulating presynaptic striatal dopamine function, either directly or through upstream effects. [6]
Concept of Salience
Usually, dopamine’s role is to mediate motivational salience and thereby gives a person the ability to determine what stimulus grabs their attention and drives the subsequent behaviour.
The salience network consists of the Anterior Cingulate Cortex (ACC), insula and the amygdala.
Schizophrenia is associated with an aberrant attribution of salience due to dysregulated striatal dopamine transmission.
Dysregulation of the dopamine system ultimately leads to irrelevant stimuli becoming more prominent which provides a basis for psychotic phenomena such as ideas of reference, where everyday occurrences may be layered with a with a heightened sense of bizarre significance. Furthermore, this misattribution of salience can lead to paranoid behaviour and persecutory delusions.![]() [7]
[7]
A stimulus, even if initially lacking inherent salience, once paired with dopaminergic activity, maintains the ability to evoke dopaminergic activity over time.
This suggests that in psychosis, once an environmental stimulus has been highlighted by aberrant dopamine signalling, it may maintain its ability to trigger dopaminergic activity, potentially cementing its position in a delusional framework, even if the system subsequently returns to normal function. [McCutcheon, et al, 2019]
LIMITATIONS OF THE DOPAMINE HYPOTHESIS OF SCHIZOPHRENIA
Current research shows that one-third of individuals with schizophrenia do not respond to non-clozapine antipsychotics despite high levels of D2-receptor occupancy.
Furthermore, a study using tetrabenazine (used as augmentation) which depletes presynaptic dopamine was not found to be effective in augmenting a clinical response in schizophrenia. [8]
Therefore, for a significant number of patients with schizophrenia, the basis of their symptoms is either unrelated to dopaminergic dysfunction or is associated with something more than just dopamine excess.
Alternatively, this could also mean that for some patients with schizophrenia there might be a non-dopaminergic sub-type of schizophrenia.
The current dopamine hypothesis of schizophrenia does not adequately explain the cognitive and negative symptoms. Current treatments which modulate dopamine transmission have only modest effects in improving these symptoms.
It has taken two decades for the dopamine hypothesis to evolve and reach its current state. More recent evidence shows another neurotransmitter, glutamate playing an essential role in schizophrenia.
The future likely holds a lot more secrets about schizophrenia which should unravel with the advances in understanding the brain.
Learn more:
Simplified Guide to Mechanisms of Action of Oral Antipsychotics
QUIZ
Loading The Dopamine Hypothesis of Schizophrenia – Advances in Neurobiology and Clinical Application
RECOMMENDED BOOKS
Dopamine theory of schizophrenia
dopamine (she or catecholamine) hypothesis pays special attention attention to dopaminergic activity in the mesolimbic brain paths.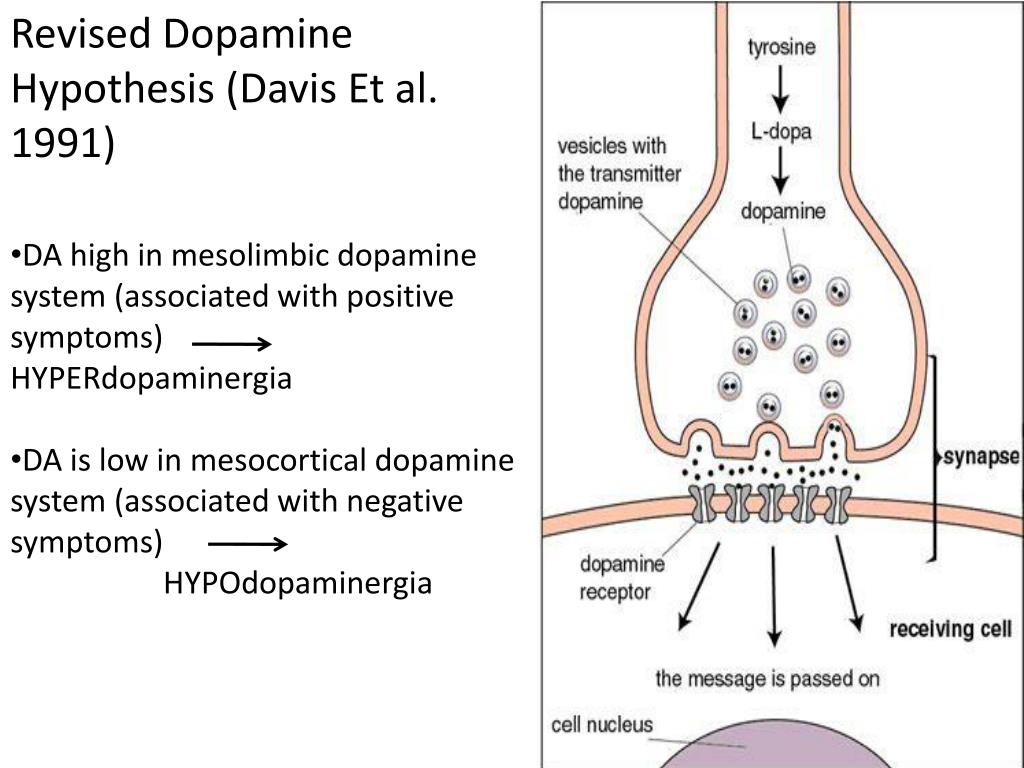
Has been put forward the so-called "dopamine theory" schizophrenia" or "dopamine hypothesis"; according to one of its versions, patients schizophrenia learn to receive pleasure, concentrating on thoughts, causing the release of dopamine and overtax their "system" encouragement”, damage to which and cause symptoms of the disease. Among supporters of the "dopamine hypothesis" There are several different streams but in general, it connects productive symptoms of schizophrenia with disturbances in dopamine systems brain. The "dopamine theory" was very popular, but its influence in our time weakened, now many psychiatrists and schizophrenia researchers do not support this theory, considering it too simplified and unable to fully explain schizophrenia. This revision is partly contributed to the emergence new ("atypical") antipsychotics, which, when similar to the old drugs efficiency have a different spectrum effects on neurotransmitter receptors. nine0003
Primary defect dopaminergic transmission in schizophrenia could not be installed because functional assessment of dopaminergic system researchers received various results. Determination results levels of dopamine and its metabolites in blood, urine and cerebrospinal liquids turned out to be inconclusive due to a large amount of these biological environment, which leveled the possible changes associated with limited dysfunction of the dopaminergic system.
Determination results levels of dopamine and its metabolites in blood, urine and cerebrospinal liquids turned out to be inconclusive due to a large amount of these biological environment, which leveled the possible changes associated with limited dysfunction of the dopaminergic system.
Numerous attempts to confirm this hypothesis before were aimed at determining in the cerebrospinal fluid of patients main product of dopamine metabolism - Homovanillic acid. However the vast majority of researchers failed to find significant the more specific changes in the content of homovanic acid in cerebrospinal fluid of patients. nine0003
Consideration schizophrenia as a disease associated with dysregulation in dopamine system required to measure the activity dopamine-p-hydroxylase enzyme, converting dopamine to norepinephrine. Decreased activity of this key enzyme in the brain tissue of patients schizophrenia may be the cause accumulation of dopamine and a decrease in the level norepinephrine in tissues. Such data could significantly confirm dopamine the schizophrenia hypothesis. It's an assumption checked in studies of the level dopamine p-hydroxylase in the spinal cord fluids of patients and study autopsy material (brain tissue). The content and activity of dopamine-(3-hydroxylase had no significant differences in compared with control studies. nine0003
Such data could significantly confirm dopamine the schizophrenia hypothesis. It's an assumption checked in studies of the level dopamine p-hydroxylase in the spinal cord fluids of patients and study autopsy material (brain tissue). The content and activity of dopamine-(3-hydroxylase had no significant differences in compared with control studies. nine0003
Study results activity of these enzymes and the corresponding substrates in peripheral blood patients do not bring us closer to understanding the role of the dopaminergic systems of the brain in the pathogenesis of psychosis. The fact is that fluctuations in activity and the level of specific dopamine system enzymes like dopamine itself, on the periphery is not reflect the states of the same systems on brain level. Moreover, changes levels of dopamine activity in the brain get a physiological expression only when they occur strictly defined brain structures (striatum region, limbic system). As a result, the development of dopamine hypothesis has methodological limitations and cannot follow the path of measurement content of dopamine and related compounds in peripheral blood and urine of the mentally ill. nine0003
nine0003
In a few work on a posthumously taken brain tissues of patients tried to study the condition dopamine system. Was established dopamine hypersensitivity receptors that are affinity for 3N-apomorphine, in the limbic region and striatum of the brain of patients with schizophrenia. However, strong evidence is needed. that this hypersensitivity (increase in the number of receptors) is not a consequence of drug induction, i.e. not caused by chronic administration of psychotropic compounds examined patients. nine0003
Some researchers have tried to confirm dopamine hypothesis of schizophrenia by measurement of the hormone prolactin in the blood plasma of patients before and during treatment with neuroleptics. Selection prolactin from the pituitary gland is regulated dopamine system of the brain, hyperactivity which should lead to increase in its content in the blood. However noticeable changes in prolactin levels in patients not treated with psychotropic drugs were not noted, and the examination treated patients gave inconclusive and conflicting results. nine0003
nine0003
Thus, a number pharmacological and biochemical data points to a link between development mental disorders and change functions of the dopamine system in the brain synaptic and receptor levels. However, indirect methods of checking dopamine hypothesis of schizophrenia did not give positive results. Tem However, all of these approaches can be insufficiently adequate to study mechanisms of dopamine disruption brain systems. For example, if psychosis-causing changes in dopamine activities are localized only in such isolated brain structures limbic region, then all modern methods for determining this activity in biological fluids (even in cerebrospinal fluid) will be unsuitable for proof fact. Acceptability of the dopamine hypothesis to explain the nature of schizophrenia finally established with the advent more sensitive methods and adequate approaches to the study of chemical disorders at the level of the human brain. nine0003
a survival guide for those who often do not see the white line"
Approximately 45 million people worldwide suffer from bipolar disorder. It manifests itself in extreme mood swings (from euphoria to depression) and seriously complicates life. Between 25 and 50 percent of people with bipolar disorder have attempted suicide. The causes and mechanisms of development of bipolar disorder are not yet very clear to scientists, but doctors already understand how to help people with it live a normal life. In the book "Bipolar Disorder: A Survival Guide for Those Who Often Do Not See the White Stripe" (published by "AST"), journalist Maria Pushkina and psychiatrist Evgeny Kasyanov tell how bipolar disorder looks from the inside and from the outside, as well as what can be done to maintaining a balance between extreme emotional states. We invite you to read a fragment about what science thinks about the causes of bipolar disorder. nine0003
It manifests itself in extreme mood swings (from euphoria to depression) and seriously complicates life. Between 25 and 50 percent of people with bipolar disorder have attempted suicide. The causes and mechanisms of development of bipolar disorder are not yet very clear to scientists, but doctors already understand how to help people with it live a normal life. In the book "Bipolar Disorder: A Survival Guide for Those Who Often Do Not See the White Stripe" (published by "AST"), journalist Maria Pushkina and psychiatrist Evgeny Kasyanov tell how bipolar disorder looks from the inside and from the outside, as well as what can be done to maintaining a balance between extreme emotional states. We invite you to read a fragment about what science thinks about the causes of bipolar disorder. nine0003
It's all about dopamine
Traditionally, scientists explained mania and depression by an imbalance in the systems of neurotransmitters 1 : serotonin, norepinephrine and dopamine.
However, in practice, everything turned out to be more complicated.
The invention of antidepressants that act on serotonin and norepinephrine made a splash. They perfectly leveled the mood of patients with "typical" unipolar depression. This discovery was later rightly called the psychopharmacological revolution .
But when antidepressants were used to treat bipolar patients, this led to very undesirable consequences - the development of mania and mixed states. What's more, people with bipolar disorder have been found to respond better to lithium and antipsychotics, especially when they are manic.
Scientists began to understand that bipolar disorder has a slightly different nature. Over the past four decades, the main hypothesis explaining the mechanism of bipolar disorder has remained dopamine hypothesis 2 .
2
Ashok A.H.
Marques T.R.
The latest research confirms that mania is based on increased activity of dopamine receptors in the brain and subsequent hyperactivation of the "reward" system. Scientists have even managed to induce manic behavior in lab mice by artificially activating their dopamine neurons.
Scientists have even managed to induce manic behavior in lab mice by artificially activating their dopamine neurons.
Reward system is an ingenious invention of evolution. These are brain structures that "reward" us with positive emotions and a sense of pleasure for successful actions, thus literally controlling our behavior. As a result, we tend to repeat these actions over and over again.
Researchers have found that bipolar depression increases levels of a protein that transports dopamine in the striatum, the area of the brain responsible for the "reward" system. Because of this, the level of dopamine decreases, which is what we see in depression. It is the lack of dopamine that is responsible for anhedonia - the loss of the ability to enjoy what used to please you. nine0003
There is a hypothesis of hypersensitivity to reward in bipolar disorder. According to her, people who are predisposed to this disease have a particularly sensitive reward system. She reacts to the achievement of the goal and encouragement especially sharply, literally with stormy delight. Which, accordingly, encourages bipolar people to act even more actively, to strive for ever new achievements. This is how hypomanic and manic behavior occurs.
She reacts to the achievement of the goal and encouragement especially sharply, literally with stormy delight. Which, accordingly, encourages bipolar people to act even more actively, to strive for ever new achievements. This is how hypomanic and manic behavior occurs.
The hypersensitive reward system reacts with a vengeance to negative events. Therefore, the displeasure and decrease in motivation after failures in people with bipolar disorder will be stronger than in those around them. As a result, depressive symptoms develop. nine0003
However, it must be understood that the dopamine hypothesis is still only a hypothesis, that is, an assumption that is not yet sufficiently substantiated to base treatment strategies for bipolar disorder on it.
What do hormones have to do with it?
Many scientists believe that neurotransmitter imbalances in bipolar disorder are just the tip of the iceberg. There are hypotheses that explain the symptoms of other failures in the endocrine system.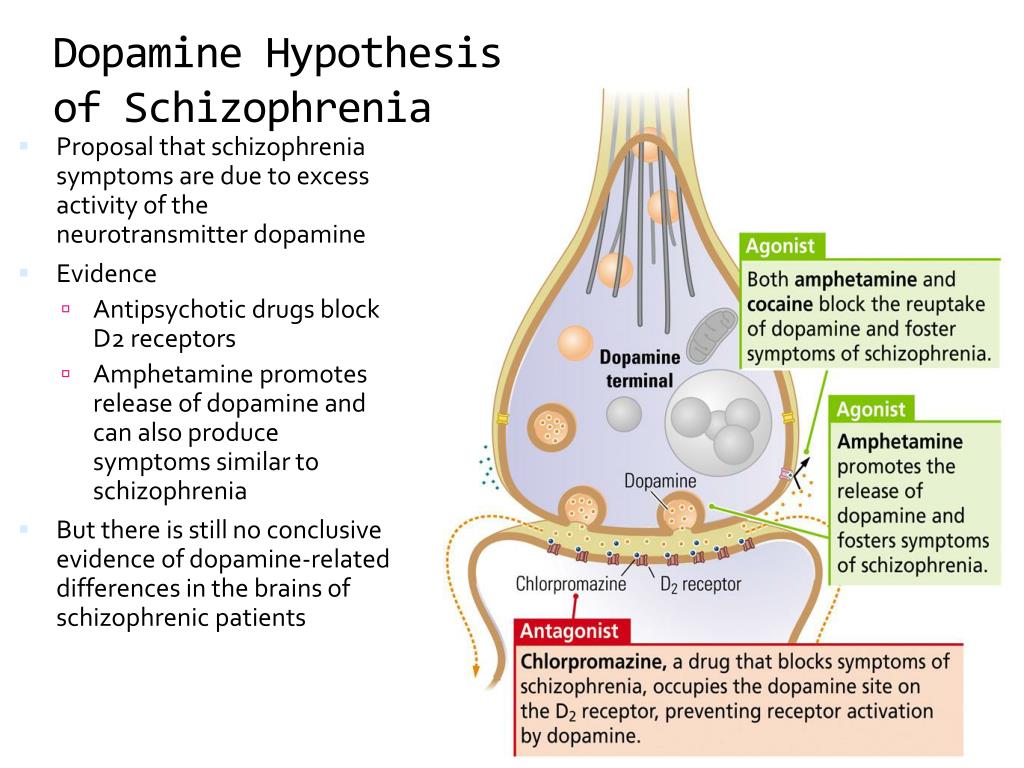 For example, violations of the "stress hormones" produced by the hypothalamus and pituitary gland, and the adrenal glands. nine0003
For example, violations of the "stress hormones" produced by the hypothalamus and pituitary gland, and the adrenal glands. nine0003
Depression is known to increase levels of corticotropin-releasing hormone and cortisol produced in the adrenal glands. Corticotropin-releasing hormone is called "the conductor of the stress symphony". It is he who triggers the mechanism of reaction to stress in the brain and then throughout the body. At the same time, the level of another stress hormone, cortisol, rises.
When the increase in the level of these hormones is short-term, the body does not suffer: evolution has adapted us to it. But if it happens for a long time (as with depression, for example), the consequences can be catastrophic: cortisol literally kills neurons in the areas of the brain responsible for thinking and memory - in the cerebral cortex and hippocampus. Perhaps this is how the weakening of cognitive abilities occurs in bipolar disorder.
However, elevated levels of stress hormones are much more common in unipolar depression than in bipolar disorder. Moreover, often in bipolar depression, the levels of stress hormones are no different from those in healthy people.
Moreover, often in bipolar depression, the levels of stress hormones are no different from those in healthy people.
Biological clock disease
Another area of research is the study of circadian rhythm disturbances (“biological clock”) in bipolar disorder.
Our brain systems that regulate circadian rhythms affect neurotransmitters - the same dopamine, serotonin and norepinephrine, the imbalance of which is associated with mood disorders. In particular, the "sleep hormone" melatonin is synthesized from serotonin at nightfall.
Mutations in circadian genes may make a person more susceptible to mood disorders, and then the person exacerbates this situation with disrupted daily routines and lack of sleep. nine0003
Everyone has probably heard that stress is bad for sleep. The fact is that the stress hormone already known to you (corticotropin-releasing hormone) suppresses the activity of all sleep centers in our brain. Evolution has thus prepared us to stay awake in times of danger.
But a depressed person experiencing chronic stress is literally deprived of sleep by this evolutionary invention.
Circadian rhythm disorders can also explain the dependence of bipolar episodes on seasons. According to a study by Fellinger M. and colleagues, the number of hospitalizations due to mania in men peaked in June, and in women in September. The number of hospitalizations due to depression for women turned out to be maximum in November (no such dependence was observed in men). With a mixed state, women were most often admitted to hospitals in June (in men, the relationship of mixed episodes with the seasons was also not found). nine0003
For information on how to restore circadian rhythms and beat insomnia, read the Bipolar Life Basics section.
BAD and hunger
Another interesting observation is related to leptin, a hormone produced by adipose tissue. Leptin is responsible for feeling full. Its synthesis is also associated with circadian rhythms. Abnormal levels of this hormone may explain the increased appetite in atypical depression.
Abnormal levels of this hormone may explain the increased appetite in atypical depression.
As we can see, there are still more blank spots in the neurobiology of bipolar disorder than clear answers. Science, of course, does not stop there. Current research into affective disorders is focused on synapses (connections between neurons) and plasticity (that is, the ability to change) in brain regions involved in the regulation of emotions, memory, sleep, and even smell - the prefrontal cortex, the hippocampus, and the amygdala. Perhaps over time they will answer the question of what exactly is wrong with the bipolar brain. nine0003
Abnormal brain
Until the middle of the 20th century, doctors did not have safe tools for studying the brain, and therefore one could only guess about the causes of manias and depressions. Post-mortem autopsies showed no brain anomalies.
Modern science has high-tech methods of neuroimaging - that is, the study of the structure and functions of the brain using images.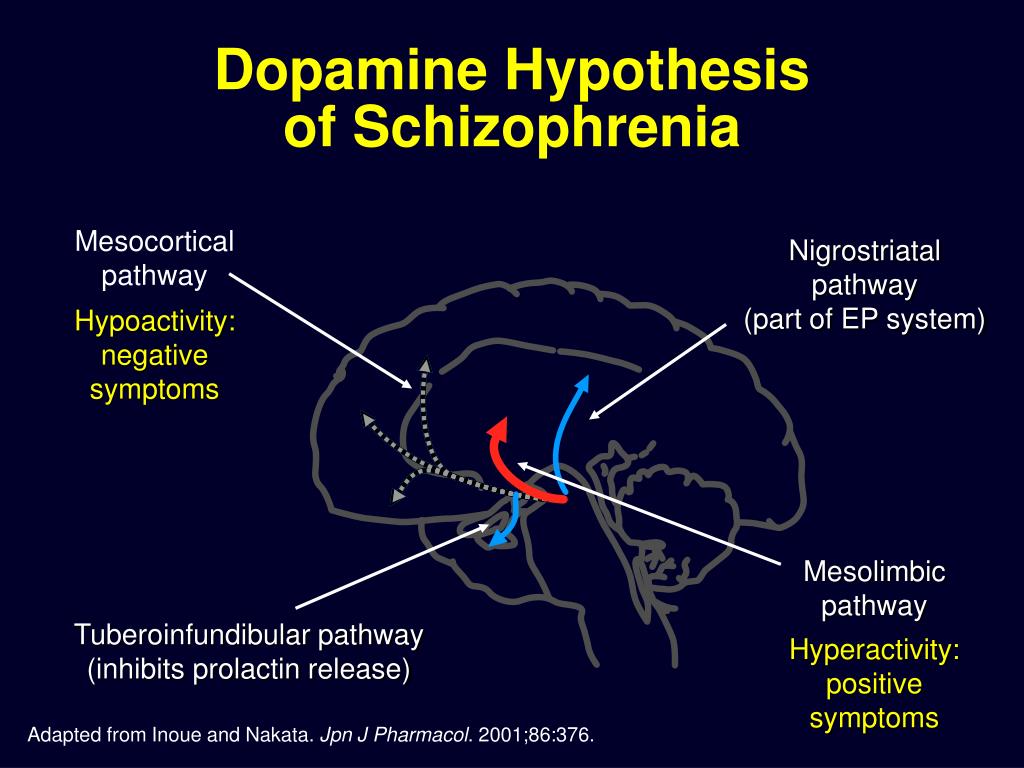 Thanks to such methods as MRI, CT, PET 3 , it was possible to detect certain disorders in the structure of the brain of bipolar people. nine0003
Thanks to such methods as MRI, CT, PET 3 , it was possible to detect certain disorders in the structure of the brain of bipolar people. nine0003
The ENIGMA Bipolar Working Group 4 reviewed over 6,500 MRI scans (the largest MRI scan of the gray matter to date!) and found the following abnormalities:
4
900.54
People with bipolar disorder had less gray matter in the frontal, temporal and parietal areas of both hemispheres of the brain.
Gray matter is the operational center of the body. It allows us to think, move, perceive the world around us. nine0003
The areas most severely affected were the left frontal operculum (the area between the anterior ascending branch of the lateral sulcus and the inferior part of the precentral sulcus), the left fusiform gyrus, and the left median frontal cortex. This phenomenon is called cortical thinning .
It has been observed that the longer the disease lasted, the thinner the cortex in the frontal, medial parietal and occipital areas of the brain.