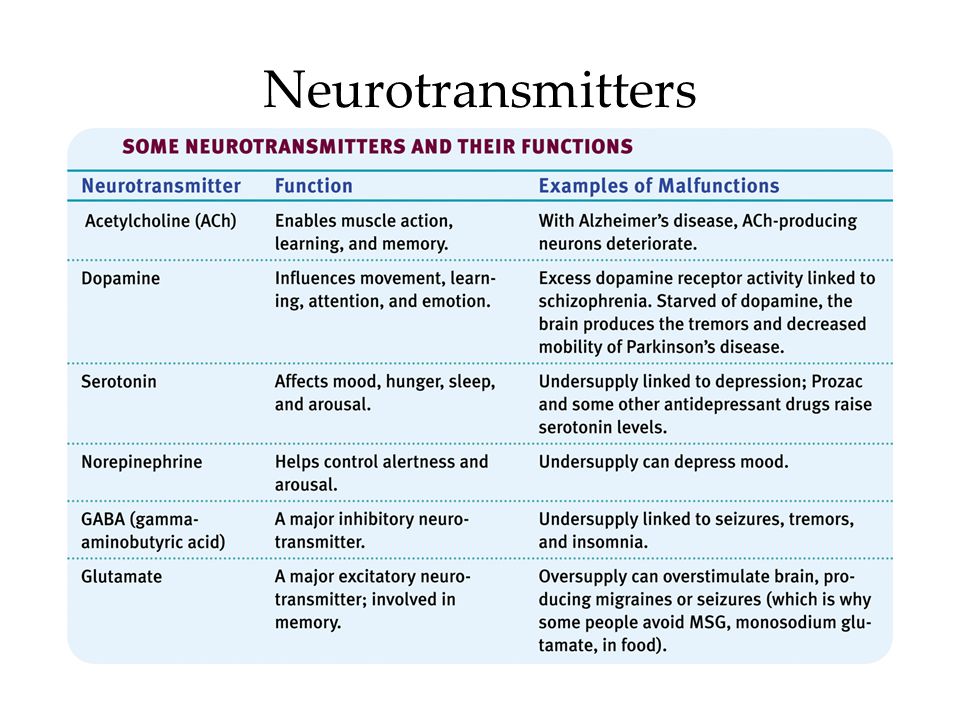
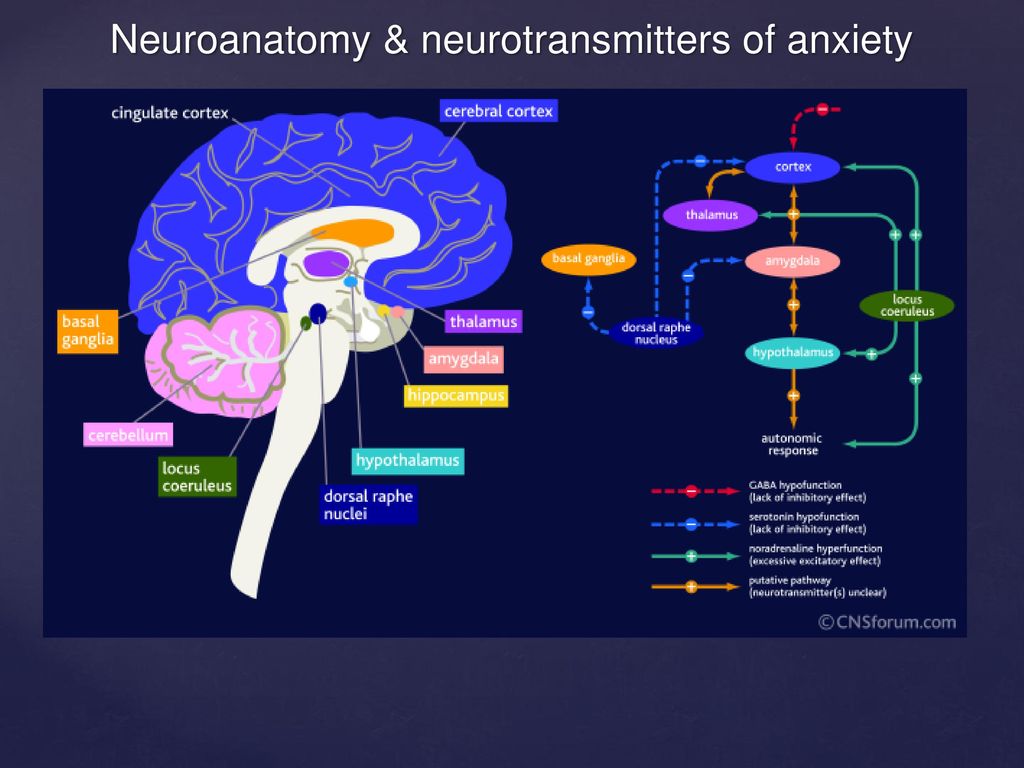
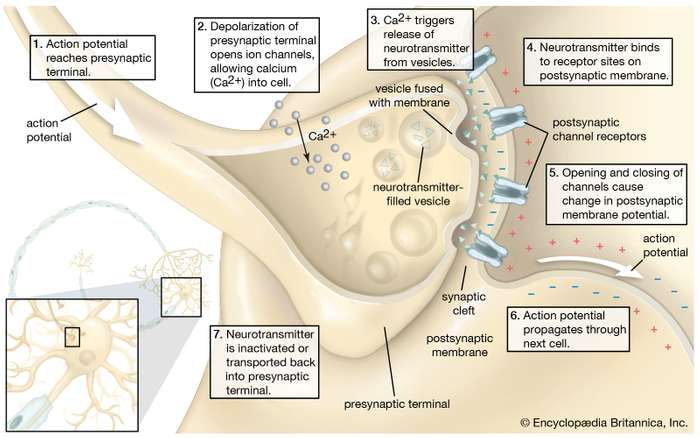
Depression neurotransmitters involved
SAMHSA’s National Helpline | SAMHSA
Your browser is not supported
Switch to Chrome, Edge, Firefox or Safari
Main page content
-
SAMHSA’s National Helpline is a free, confidential, 24/7, 365-day-a-year treatment referral and information service (in English and Spanish) for individuals and families facing mental and/or substance use disorders.
Also visit the online treatment locator.
SAMHSA’s National Helpline, 1-800-662-HELP (4357) (also known as the Treatment Referral Routing Service), or TTY: 1-800-487-4889 is a confidential, free, 24-hour-a-day, 365-day-a-year, information service, in English and Spanish, for individuals and family members facing mental and/or substance use disorders.
This service provides referrals to local treatment facilities, support groups, and community-based organizations.
Also visit the online treatment locator, or send your zip code via text message: 435748 (HELP4U) to find help near you. Read more about the HELP4U text messaging service.
The service is open 24/7, 365 days a year.
English and Spanish are available if you select the option to speak with a national representative. Currently, the 435748 (HELP4U) text messaging service is only available in English.
In 2020, the Helpline received 833,598 calls. This is a 27 percent increase from 2019, when the Helpline received a total of 656,953 calls for the year.
The referral service is free of charge. If you have no insurance or are underinsured, we will refer you to your state office, which is responsible for state-funded treatment programs. In addition, we can often refer you to facilities that charge on a sliding fee scale or accept Medicare or Medicaid. If you have health insurance, you are encouraged to contact your insurer for a list of participating health care providers and facilities.
In addition, we can often refer you to facilities that charge on a sliding fee scale or accept Medicare or Medicaid. If you have health insurance, you are encouraged to contact your insurer for a list of participating health care providers and facilities.
The service is confidential. We will not ask you for any personal information. We may ask for your zip code or other pertinent geographic information in order to track calls being routed to other offices or to accurately identify the local resources appropriate to your needs.
No, we do not provide counseling. Trained information specialists answer calls, transfer callers to state services or other appropriate intake centers in their states, and connect them with local assistance and support.
-
Suggested Resources
What Is Substance Abuse Treatment? A Booklet for Families
Created for family members of people with alcohol abuse or drug abuse problems.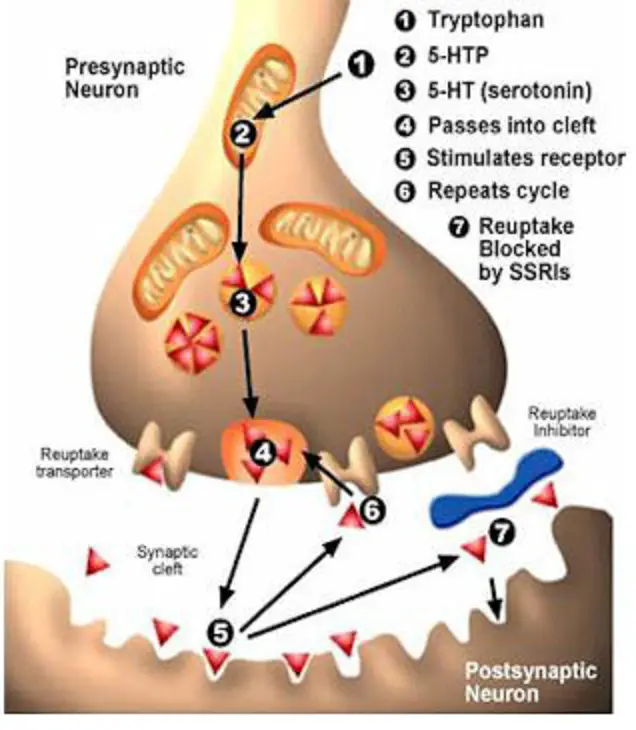 Answers questions about substance abuse, its symptoms, different types of treatment, and recovery. Addresses concerns of children of parents with substance use/abuse problems.
Answers questions about substance abuse, its symptoms, different types of treatment, and recovery. Addresses concerns of children of parents with substance use/abuse problems.It's Not Your Fault (NACoA) (PDF | 12 KB)
Assures teens with parents who abuse alcohol or drugs that, "It's not your fault!" and that they are not alone. Encourages teens to seek emotional support from other adults, school counselors, and youth support groups such as Alateen, and provides a resource list.After an Attempt: A Guide for Taking Care of Your Family Member After Treatment in the Emergency Department
Aids family members in coping with the aftermath of a relative's suicide attempt. Describes the emergency department treatment process, lists questions to ask about follow-up treatment, and describes how to reduce risk and ensure safety at home.Family Therapy Can Help: For People in Recovery From Mental Illness or Addiction
Explores the role of family therapy in recovery from mental illness or substance abuse. Explains how family therapy sessions are run and who conducts them, describes a typical session, and provides information on its effectiveness in recovery.
Explains how family therapy sessions are run and who conducts them, describes a typical session, and provides information on its effectiveness in recovery.For additional resources, please visit the SAMHSA Store.
Last Updated: 08/30/2022
Alcohol, Tobacco, and Other Drugs
Your browser is not supported
Switch to Chrome, Edge, Firefox or Safari
Misusing alcohol, tobacco, and other drugs can have both immediate and long-term health effects.The misuse and abuse of alcohol, tobacco, illicit drugs, and prescription medications affect the health and well-being of millions of Americans. NSDUH estimates allow researchers, clinicians, policymakers, and the general public to better understand and improve the nation’s behavioral health. These reports and detailed tables present estimates from the 2021 National Survey on Drug Use and Health (NSDUH).
Alcohol
Data:
- Among the 133.1 million current alcohol users aged 12 or older in 2021, 60.0 million people (or 45.1%) were past month binge drinkers. The percentage of people who were past month binge drinkers was highest among young adults aged 18 to 25 (29.2% or 9.8 million people), followed by adults aged 26 or older (22.4% or 49.3 million people), then by adolescents aged 12 to 17 (3.8% or 995,000 people). (2021 NSDUH)
- Among people aged 12 to 20 in 2021, 15.1% (or 5.9 million people) were past month alcohol users. Estimates of binge alcohol use and heavy alcohol use in the past month among underage people were 8.3% (or 3.2 million people) and 1.6% (or 613,000 people), respectively. (2021 NSDUH)
- In 2020, 50.0% of people aged 12 or older (or 138.5 million people) used alcohol in the past month (i.e., current alcohol users) (2020 NSDUH)
- Among the 138.5 million people who were current alcohol users, 61.6 million people (or 44.
 4%) were classified as binge drinkers and 17.7 million people (28.8% of current binge drinkers and 12.8% of current alcohol users) were classified as heavy drinkers (2020 NSDUH)
4%) were classified as binge drinkers and 17.7 million people (28.8% of current binge drinkers and 12.8% of current alcohol users) were classified as heavy drinkers (2020 NSDUH) - The percentage of people who were past month binge alcohol users was highest among young adults aged 18 to 25 (31.4%) compared with 22.9% of adults aged 26 or older and 4.1% of adolescents aged 12 to 17 (2020 NSDUH)
- Excessive alcohol use can increase a person’s risk of stroke, liver cirrhosis, alcoholic hepatitis, cancer, and other serious health conditions
- Excessive alcohol use can also lead to risk-taking behavior, including driving while impaired. The Centers for Disease Control and Prevention reports that 29 people in the United States die in motor vehicle crashes that involve an alcohol-impaired driver daily
Programs/Initiatives:
- STOP Underage Drinking interagency portal - Interagency Coordinating Committee on the Prevention of Underage Drinking
- Interagency Coordinating Committee on the Prevention of Underage Drinking
- Talk.
 They Hear You.
They Hear You. - Underage Drinking: Myths vs. Facts
- Talking with your College-Bound Young Adult About Alcohol
Relevant links:
- National Association of State Alcohol and Drug Abuse Directors
- Department of Transportation Office of Drug & Alcohol Policy & Compliance
- Alcohol Policy Information Systems Database (APIS)
- National Institute on Alcohol Abuse and Alcoholism
Tobacco
Data:
- In 2020, 20.7% of people aged 12 or older (or 57.3 million people) used nicotine products (i.e., used tobacco products or vaped nicotine) in the past month (2020 NSDUH)
- Among past month users of nicotine products, nearly two thirds of adolescents aged 12 to 17 (63.1%) vaped nicotine but did not use tobacco products. In contrast, 88.9% of past month nicotine product users aged 26 or older used only tobacco products (2020 NSDUH)
- Tobacco use is the leading cause of preventable death, often leading to lung cancer, respiratory disorders, heart disease, stroke, and other serious illnesses.
 The CDC reports that cigarette smoking causes more than 480,000 deaths each year in the United States
The CDC reports that cigarette smoking causes more than 480,000 deaths each year in the United States - The CDC’s Office on Smoking and Health reports that more than 16 million Americans are living with a disease caused by smoking cigarettes
Electronic cigarette (e-cigarette) use data:
- In 2021, 13.2 million people aged 12 or older (or 4.7%) used an e-cigarette or other vaping device to vape nicotine in the past month. The percentage of people who vaped nicotine was highest among young adults aged 18 to 25 (14.1% or 4.7 million people), followed by adolescents aged 12 to 17 (5.2% or 1.4 million people), then by adults aged 26 or older (3.2% or 7.1 million people).
- Among people aged 12 to 20 in 2021, 11.0% (or 4.3 million people) used tobacco products or used an e-cigarette or other vaping device to vape nicotine in the past month. Among people in this age group, 8.1% (or 3.1 million people) vaped nicotine, 5.4% (or 2.1 million people) used tobacco products, and 3.
 4% (or 1.3 million people) smoked cigarettes in the past month. (2021 NSDUH)
4% (or 1.3 million people) smoked cigarettes in the past month. (2021 NSDUH) - Data from the Centers for Disease Control and Prevention’s 2020 National Youth Tobacco Survey. Among both middle and high school students, current use of e-cigarettes declined from 2019 to 2020, reversing previous trends and returning current e-cigarette use to levels similar to those observed in 2018
- E-cigarettes are not safe for youth, young adults, or pregnant women, especially because they contain nicotine and other chemicals
Resources:
- Tips for Teens: Tobacco
- Tips for Teens: E-cigarettes
- Implementing Tobacco Cessation Programs in Substance Use Disorder Treatment Settings
- Synar Amendment Program
Links:
- Truth Initiative
- FDA Center for Tobacco Products
- CDC Office on Smoking and Health
- National Institute on Drug Abuse: Tobacco, Nicotine, and E-Cigarettes
- National Institute on Drug Abuse: E-Cigarettes
Opioids
Data:
- Among people aged 12 or older in 2021, 3.
 3% (or 9.2 million people) misused opioids (heroin or prescription pain relievers) in the past year. Among the 9.2 million people who misused opioids in the past year, 8.7 million people misused prescription pain relievers compared with 1.1 million people who used heroin. These numbers include 574,000 people who both misused prescription pain relievers and used heroin in the past year. (2021 NSDUH)
3% (or 9.2 million people) misused opioids (heroin or prescription pain relievers) in the past year. Among the 9.2 million people who misused opioids in the past year, 8.7 million people misused prescription pain relievers compared with 1.1 million people who used heroin. These numbers include 574,000 people who both misused prescription pain relievers and used heroin in the past year. (2021 NSDUH) - Among people aged 12 or older in 2020, 3.4% (or 9.5 million people) misused opioids in the past year. Among the 9.5 million people who misused opioids in the past year, 9.3 million people misused prescription pain relievers and 902,000 people used heroin (2020 NSDUH)
- According to the Centers for Disease Control and Prevention’s Understanding the Epidemic, an average of 128 Americans die every day from an opioid overdose
Resources:
- Medications for Substance Use Disorders
- Opioid Overdose Prevention Toolkit
- TIP 63: Medications for Opioid Use Disorder
- Use of Medication-Assisted Treatment for Opioid Use Disorder in Criminal Justice Settings
- Opioid Use Disorder and Pregnancy
- Clinical Guidance for Treating Pregnant and Parenting Women With Opioid Use Disorder and Their Infants
- The Facts about Buprenorphine for Treatment of Opioid Addiction
- Pregnancy Planning for Women Being Treated for Opioid Use Disorder
- Tips for Teens: Opioids
- Rural Opioid Technical Assistance Grants
- Tribal Opioid Response Grants
- Provider’s Clinical Support System - Medication Assisted Treatment Grant Program
Links:
- National Institute on Drug Abuse: Opioids
- National Institute on Drug Abuse: Heroin
- HHS Prevent Opioid Abuse
- Community Anti-Drug Coalitions of America
- Addiction Technology Transfer Center (ATTC) Network
- Prevention Technology Transfer Center (PTTC) Network
Marijuana
Data:
- In 2021, marijuana was the most commonly used illicit drug, with 18.
 7% of people aged 12 or older (or 52.5 million people) using it in the past year. The percentage was highest among young adults aged 18 to 25 (35.4% or 11.8 million people), followed by adults aged 26 or older (17.2% or 37.9 million people), then by adolescents aged 12 to 17 (10.5% or 2.7 million people).
7% of people aged 12 or older (or 52.5 million people) using it in the past year. The percentage was highest among young adults aged 18 to 25 (35.4% or 11.8 million people), followed by adults aged 26 or older (17.2% or 37.9 million people), then by adolescents aged 12 to 17 (10.5% or 2.7 million people). - The percentage of people who used marijuana in the past year was highest among young adults aged 18 to 25 (34.5%) compared with 16.3% of adults aged 26 or older and 10.1% of adolescents aged 12 to 17 (2020 NSDUH)
- Marijuana can impair judgment and distort perception in the short term and can lead to memory impairment in the long term
- Marijuana can have significant health effects on youth and pregnant women.
Resources:
- Know the Risks of Marijuana
- Marijuana and Pregnancy
- Tips for Teens: Marijuana
Relevant links:
- National Institute on Drug Abuse: Marijuana
- Addiction Technology Transfer Centers on Marijuana
- CDC Marijuana and Public Health
Emerging Trends in Substance Misuse:
- Methamphetamine—In 2019, NSDUH data show that approximately 2 million people used methamphetamine in the past year.
 Approximately 1 million people had a methamphetamine use disorder, which was higher than the percentage in 2016, but similar to the percentages in 2015 and 2018. The National Institute on Drug Abuse Data shows that overdose death rates involving methamphetamine have quadrupled from 2011 to 2017. Frequent meth use is associated with mood disturbances, hallucinations, and paranoia.
Approximately 1 million people had a methamphetamine use disorder, which was higher than the percentage in 2016, but similar to the percentages in 2015 and 2018. The National Institute on Drug Abuse Data shows that overdose death rates involving methamphetamine have quadrupled from 2011 to 2017. Frequent meth use is associated with mood disturbances, hallucinations, and paranoia. - Cocaine—In 2019, NSDUH data show an estimated 5.5 million people aged 12 or older were past users of cocaine, including about 778,000 users of crack. The CDC reports that overdose deaths involving have increased by one-third from 2016 to 2017. In the short term, cocaine use can result in increased blood pressure, restlessness, and irritability. In the long term, severe medical complications of cocaine use include heart attacks, seizures, and abdominal pain.
- Kratom—In 2019, NSDUH data show that about 825,000 people had used Kratom in the past month. Kratom is a tropical plant that grows naturally in Southeast Asia with leaves that can have psychotropic effects by affecting opioid brain receptors.
 It is currently unregulated and has risk of abuse and dependence. The National Institute on Drug Abuse reports that health effects of Kratom can include nausea, itching, seizures, and hallucinations.
It is currently unregulated and has risk of abuse and dependence. The National Institute on Drug Abuse reports that health effects of Kratom can include nausea, itching, seizures, and hallucinations.
Resources:
- Tips for Teens: Methamphetamine
- Tips for Teens: Cocaine
- National Institute on Drug Abuse
More SAMHSA publications on substance use prevention and treatment.
Last Updated: 03/22/2023
Neurobiology of depression: serotonin system of the brain
Major depression is a common mental disorder that is one of the most common causes of disability [1, 2]. This disease occurs in all age groups and affects people of both sexes in every region of the world. The experience of the last decades has shown that the prospects for studying depression are related to its neurobiology. The molecular hypothesis is widely used to explain the pathogenetic mechanisms of depression. According to the latter, adverse environmental factors such as stress affect genetic vulnerability, which causes maladaptive changes in the chain of neurotransmitters, among which monoamines play a major role.![]() In most of the existing achievements in the treatment of the disease, the effects on the deciphered mediator mechanisms of pathogenesis are also realized [3].
In most of the existing achievements in the treatment of the disease, the effects on the deciphered mediator mechanisms of pathogenesis are also realized [3].
One of the most important systems of cerebral neurotransmission involved in the pathogenesis of depression is the serotonin system. This neurotransmitter system has a long evolutionary history and is involved in a number of behavioral acts and emotional manifestations [4]. It is the subject of a significant number of studies, a review of which is presented in this publication.
To better understand the integration of the serotonin system into the brain's mood regulation processes, the available data on the influence of different cerebral regions on affective manifestations should first be considered. Thus, executive functions, including the modulation of emotional behavior, which may be related to the formation of cognitive symptoms of depression (depressive vision of the future), are associated with hypoactivation of the left frontal cortex [5].
The emotional memory system, including the amygdala and hippocampus, is also involved in the realization of manifestations of depression. Depressed patients demonstrate a predominant focus on negative past events [6]. Dysfunction of striatal circles that perform psychomotor functions can explain motor symptoms depression. Eating Disorders and violations of a number of other somatic functions indicate the involvement of the hypothalamus in the process and the hypothalamic-pituitary-adrenal axis.
These brain formations are anatomically and functionally interconnected with the help of neuronal circles [4].
Numerous experimental literature indicate the importance of the pathways that unite the frontal, paralimbic (ventral frontal cortex, cingular gyrus, insula, anterior temporal pole), striatal and stem regions into a single network in the implementation of affective and motivational processes [7-9]. In turn, with the help of functional neuroimaging methods, disturbances in the activity of the above brain regions in depressive patients were detected [10].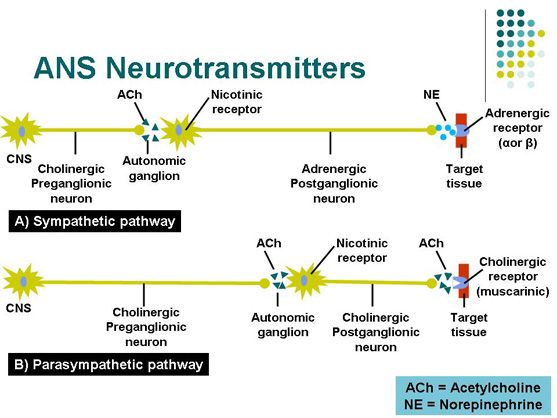 The development of a neuroanatomical model of depression was facilitated by data on the occurrence of depressive disorders in organic lesions of various brain structures. An example is ischemic lesions of the left frontal lobe in post-stroke depression [11, 12], as well as damage to the frontostriatal pathways in patients with vascular depression and Parkinson's disease [13-15].
The development of a neuroanatomical model of depression was facilitated by data on the occurrence of depressive disorders in organic lesions of various brain structures. An example is ischemic lesions of the left frontal lobe in post-stroke depression [11, 12], as well as damage to the frontostriatal pathways in patients with vascular depression and Parkinson's disease [13-15].
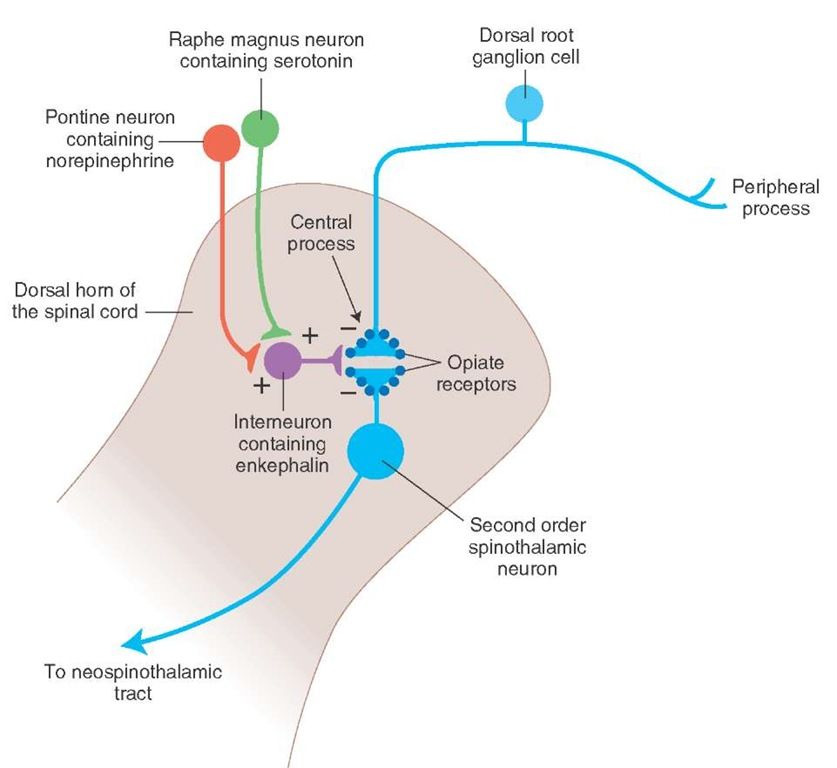
The serotonin system of the brain is an integral part of the described neuronal networks of mood regulation. Serotonergic neurons are grouped in 9 nuclei of the brainstem. Most of them coincide with the medially located raphe nucleus [16]. Serotonin (5-hydroxytryptamine [5-HT]) is synthesized in these nuclei from tryptophan.
The ascending terminals of serotonergic nuclei take part in the regulation of affective processes, which end in a large number of brain structures: subcortical formations (caudate nucleus, putamen, anterior and medial nuclei of the thalamus), diencephalon, olfactory brain and a number of formations associated with the reticular formation, cortex of large hemispheres, amygdala and hypothalamus. At the same time, there is much more serotonin in the cortex of the limbic system than in neocortical regions [17-19].
At the same time, there is much more serotonin in the cortex of the limbic system than in neocortical regions [17-19].
The importance of disruption of the serotonin synthesis pathway for the onset of depression has been shown in studies investigating the effects of dietary restriction of tryptophan intake. The hypotryptophan diet resulted in depressive symptoms in healthy individuals and in depressed patients in remission. According to positron emission tomography, the examined patients showed a decrease in the activity of the pre- and orbitofrontal cortex, as well as the thalamus [20]. There is convincing evidence of the genetic determinism of the synthesis of serotonin in the brain. It is known that the human genome contains the 5-HTT gene, whose activity regulates the level of serotonin produced by the brain [21].
Serotonin fulfills its physiological role by acting on 5-HT receptors.
Currently, more than 15 types of serotonin receptors are known [22–26], but not all of them have been identified in the human brain.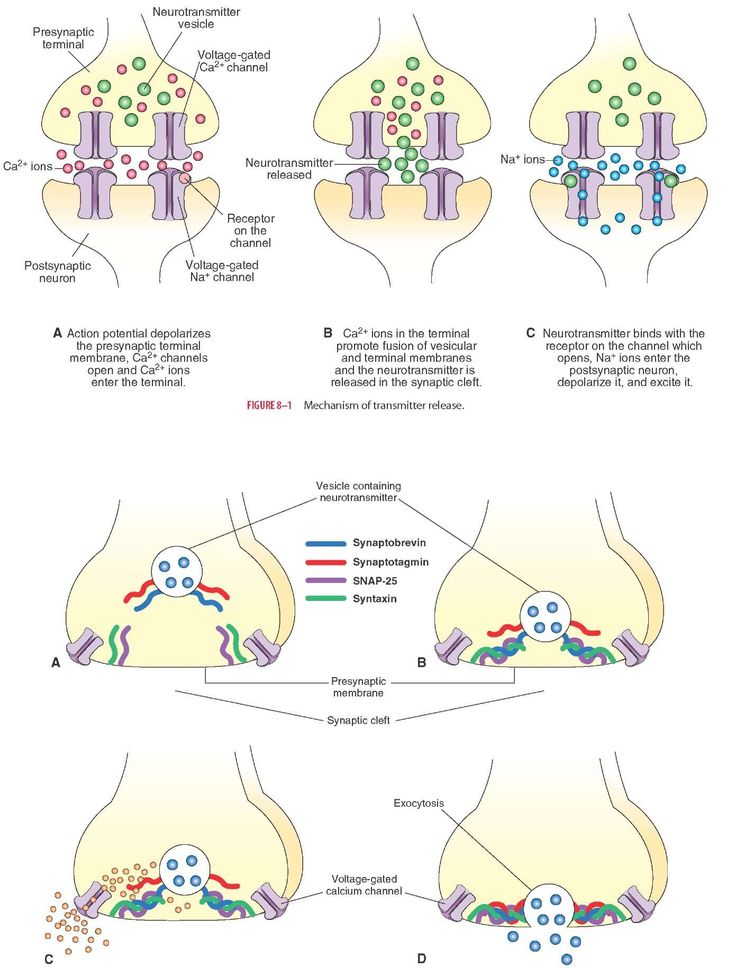
In the central nervous system (CNS) of mammals, serotonin 5-HT 1 receptors and five of their subtypes - A, B, D, E, F, are found, which are proteins containing 365-422 amino acid residues. Through inhibitory G-proteins, these receptors are coupled to adenylate cyclase, the activity of which is suppressed upon their activation.
5-HT 1A receptors are predominantly localized in the hippocampus, tonsils, septum pellucidum – structures involved in mood formation. These CNS receptors are located on the pre- and postsynaptic membranes [27]. Presynaptic 5-HT 1A receptors regulate the intensity of serotonin release from presynaptic neuronal terminals by the feedback principle. Through stimulation of postsynaptic 5-HT 1A receptors, a number of important physiological functions of serotonin are realized: mood regulation, obsessive-compulsive reactions, sexual behavior, appetite control, thermoregulation, cardiovascular regulation. It is this type of receptor that is involved in the implementation of the antidepressant effect of selective serotonin reuptake inhibitors, the antidepressant and anti-anxiety effect of buspirone.![]()
The subtype of human 5-HT 1D receptors (functional analog of rat 5-HT 1B receptors) is localized in the frontal cortex, striatum, and basal ganglia [22, 28]. Presynaptic 5-HT 1D receptors play the role of autoreceptors through which a negative feedback between the level of extra- and intraneuronal serotonin is carried out. Perhaps they also play the role of heteroreceptors, through which the release of other neurotransmitters, such as dopamine, acetylcholine, glutamate, is controlled. Stimulation of postsynaptic receptors of this subtype in experimental models caused prolonged hyperactivity, antidepressant effect, decreased pain sensitivity and appetite, and hypothermia.
It has recently been shown that the functioning of the 5-HT 1B/D receptor depends on the P11 peptide belonging to the S100 protein group. The concentration of P11 peptide in the brain of patients with depression was low. Long-term antidepressant treatment increases the level of this peptide in brain tissue [3].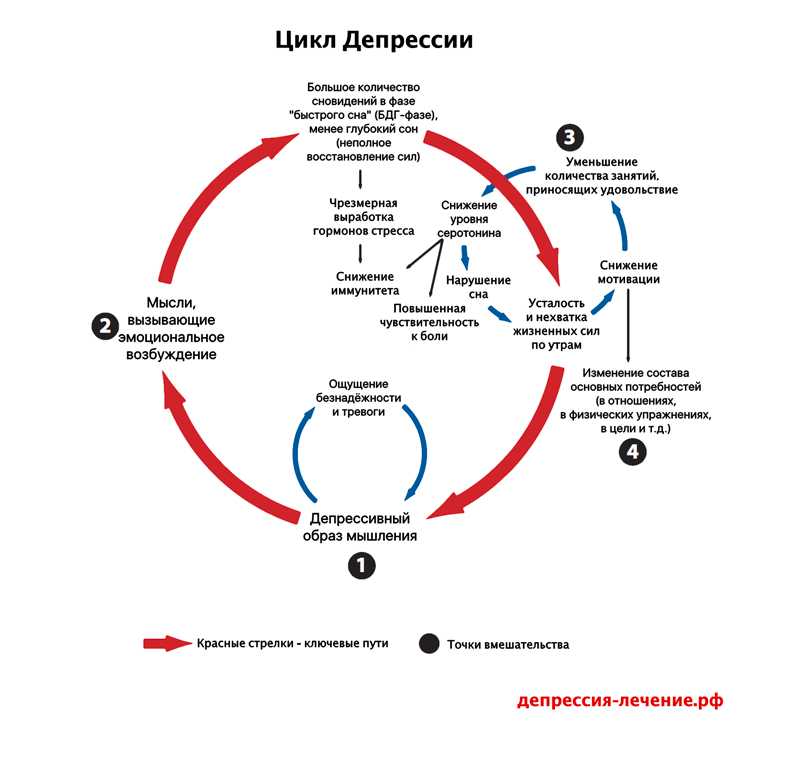 The function of other 5-HT 1 receptor subtypes has not yet been established.
The function of other 5-HT 1 receptor subtypes has not yet been established.
5-HT 2 receptors have been found in the human CNS. Their family consists of three subtypes: 5-HT 2A , 5-HT 2B , 5-HT 2C [22, 29, 30]. To a greater extent, such receptors are present in the pyramidal neurons of the frontal cortex, the putamen, and to a lesser extent, in the hippocampus and caudate nucleus. They are part of the brain reinforcement system, the low activity of which causes the occurrence of anhedonia, one of the key symptoms of depression [22]. 5-HT 2A receptors mediate the anxiogenic effect, participate in the formation of sexual behavior, and are involved in the regulation of sleep. A decrease in their number was noted in post-mortem studies in people who suffered from depression and committed suicide. 5-HT 9 activation0014 2A receptor causes an increase in concentration dopamine in the striatum. Modern atypical Antipsychotics are highly active in relation to this subtype, which is associated with the antidepressant effect of these drugs [31].![]() Antagonists of 5-HT 2A receptors increase the duration of slow-wave sleep, improving its quality, while agonists reduce the phase of fast-wave sleep.
Antagonists of 5-HT 2A receptors increase the duration of slow-wave sleep, improving its quality, while agonists reduce the phase of fast-wave sleep.
5-HT 2C CNS receptors are most abundant in the hippocampus, cerebral cortex, striatum, and substantia nigra. Agonists of these receptors cause anxiogenic and panic effects, disturb sleep. Blockade 5-HT 2C receptor is one of the mechanisms of depression treatment.
This is related to the effectiveness of antidepressants that are antagonists of these receptors (mianserin, imipramine, maprotiline, amitriptyline, desipramine, agomelatine) [32, 33]. Antagonists 5-HT 2C receptors improve sleep [22, 30] and have anxiolytic properties. The latter partly explains the anti-anxiety effect of selective serotonin reuptake inhibitors.
5-HT 3 receptors are located in the solitary tract, gelatinous substance, nuclei of the trigeminal and vagus nerves, hippocampus. Their central antagonists have an anxiolytic effect, increase cognitive abilities, change the sensitivity of nociceptive neurons, and have an antiemetic effect.
5-HT 4 receptors are maximally represented in areas saturated with dopaminergic neurons (basal nuclei, accumbens). They are localized on GABAergic and cholinergic interneurons and GABAergic projections into the substantia nigra. Agonists of these receptors can increase the activity of dopaminergic systems, while antagonists can block this effect. There are data on the anxiolytic effect of 5-HT 4 receptor antagonists [22, 34].
5-HT 6 -receptors are located in the striatum, amygdala, hippocampus, cortex, olfactory bulb. Various antidepressants (clomipramine, amitriptyline, nortriptyline, doxepin) have a high affinity for them and are their antagonists.
5-HT 7 receptors are present in the hypothalamus, thalamus, and brain stem. They can participate in the organization of circadian rhythms by influencing the suprachiasmatic nuclei. In the future, 5-HT 6 - and 5-HT 7 receptors may become a target for modeling depression [3].
The next level of disorders of the serotonin system in depression is reuptake 5-HT from the synaptic cleft to the presynaptic neuron, which is carried by the serotonin transporter protein. The density of this protein in the brain of depressed patients decreased, which was detected using functional neuroimaging methods, and in those who died due to suicide, according to postmortem histochemical studies [35].
Individual features of serotonin turnover in the CNS, among other hereditary factors, depend on the effects of the
serotonin carrier gene (5-HTT). This gene is located on the 17th chromosome. It describes several polymorphic regions, including an insertion-deletion polymorphism (5-HTTLPR) found in the promoter region and represented by two allelic variants - l (long) and s (short - with a deletion). This polymorphism is functional [36-38].
A number of authors found an association between 5-HTTLPR polymorphism and the development of depressive states in response to various stressors [39].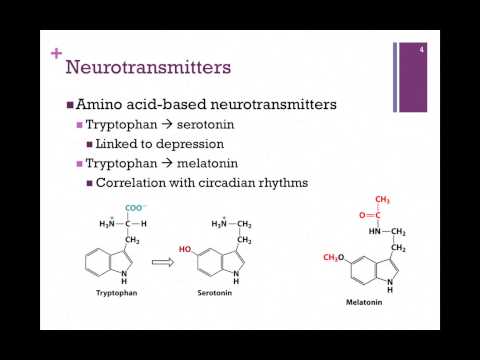 Individuals with at least one short allele in the
Individuals with at least one short allele in the
genotype exhibited more severe depressive symptoms, were more likely to be diagnosed with a DSM-IV depressive episode, and reported more suicidal thoughts and attempts during depressive episodes compared with homozygotes for the long allele. The role of the serotonin transporter gene in mediating the association between stressful life events and the subsequent development of depressive symptoms and physical distress was later confirmed by other authors [40-42]. In addition, it was found that healthy people - carriers of the short allele - are more characterized by increased emotional reactivity and anxiety, that is, personality traits that are considered as predispositional in relation to affective disorders [43, 44].
The facts described above testify to the great importance of the serotonin system for the functioning of brain areas that are directly related to the regulation of affective processes: frontal regions that modulate emotional behavior; the limbic region, which is related to emotional and cognitive impairment in depression; fronto-striatal structures that determine the occurrence of anhedonia; psychomotor disorders. Separately, it is necessary to single out the role of the serotonin system in the functioning of the hypothalamic region - the most important link in neuro-endocrine, autonomic, circadian regulation.
Separately, it is necessary to single out the role of the serotonin system in the functioning of the hypothalamic region - the most important link in neuro-endocrine, autonomic, circadian regulation.
Serotonin dysfunction directly affects limbic-hypothalamic-pituitary-adrenal regulation in patients with depression [45]. Depression is associated with increased daily production of adrenocorticotropic hormone. His hyperproduction can be explained by an increase in the production of corticotropin-releasing factor, the synthesis of which is normally limited by the feedback mechanism by the level of cortisol in the blood plasma.
Violation of the inhibitory effects of cortisol on the production of corticotropin-releasing factor in depression is associated with impaired function of glucocorticoid and 5-HT 1A receptors. The result of hyperactivity hypothalamic-pituitary-adrenal axis in patients with depression is an increase in plasma cortisol levels. Hypercortisolemia, in turn, leads to a decrease in the activity of postsynaptic 5-HT 1A receptors, one of the main manifestations of serotonin dysfunction.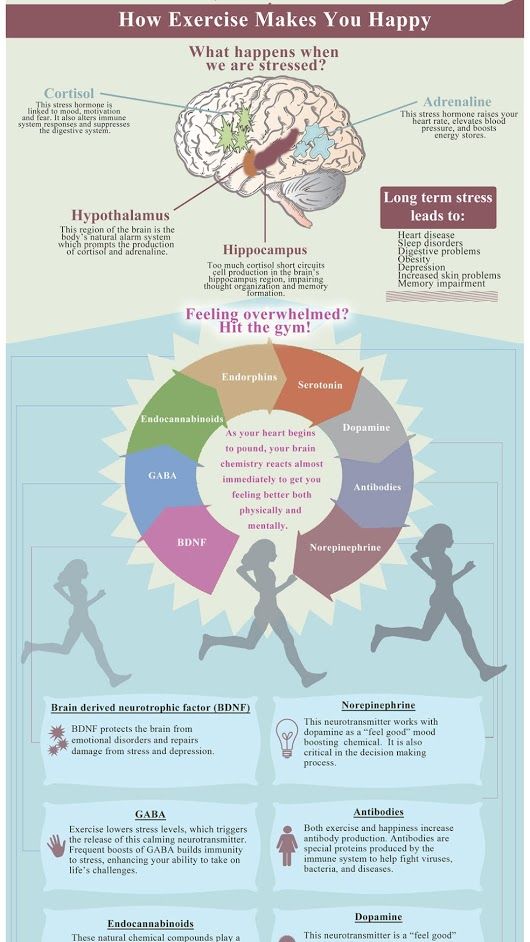 Thus, a vicious circle is closed.
Thus, a vicious circle is closed.
Cortisol also potentiates an increase in adrenaline production. This is associated with an increase in the activity of the sympathetic link of the segmental division of the autonomic nervous system. These mechanisms are responsible for many autonomic symptoms of depression.
The serotonergic system is involved in the regulation of the sleep-wake cycle. Not surprisingly, one of the most common symptoms of depression is sleep disturbance. It is believed that the main generator of circadian rhythms, localized in the suprachiasmatic nucleus of the anterior hypothalamus [46], receives information about the level of body activity from the raphe nuclei along with stimuli from the intergeniculate nuclei of the lateral geniculate body [47, 48]. Blockade 5-HT 2C receptors in the hypothalamic region, which become hypersensitive in depression, according to Krauchi et al. (1997) and Leproult et al. (2005), can resynchronize the circadian rhythm and induce antidepressant effects [3].
Effects on serotonin neurotransmission are implemented in the mechanisms of action of many modern antidepressants and other psychotropic drugs. For some drugs, these mechanisms are the main pharmacodynamic effect, for others they are of additional importance.
Inhibition of serotonin reuptake underlies the pharmacodynamics of a large number of antidepressants: selective serotonin reuptake inhibitors (SSRIs), serotonin and norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants (TCAs).
SSRIs (citalopram, sertraline, fluoxetine, fluvoxamine, paroxetine) act on the main site of the serotonin transporter protein. Escitalopram blocks both the basic and allosteric sites of this protein. Blockade of the serotonin transporter protein causes an initial increase in the concentration of 5-HT in the somatodendritic zone (but not in the zone of the axonal terminal). This, in turn, causes a decrease in the activity of 5-HT 1A autoreceptors. Since their role is to suppress impulses coming to serotonergic neurons, as well as to suppress the synthesis and release of serotonin, the blockade of receptors causes the release of neurons from inhibitory influences and increases the release of serotonin from the axonal terminal into the synaptic cleft.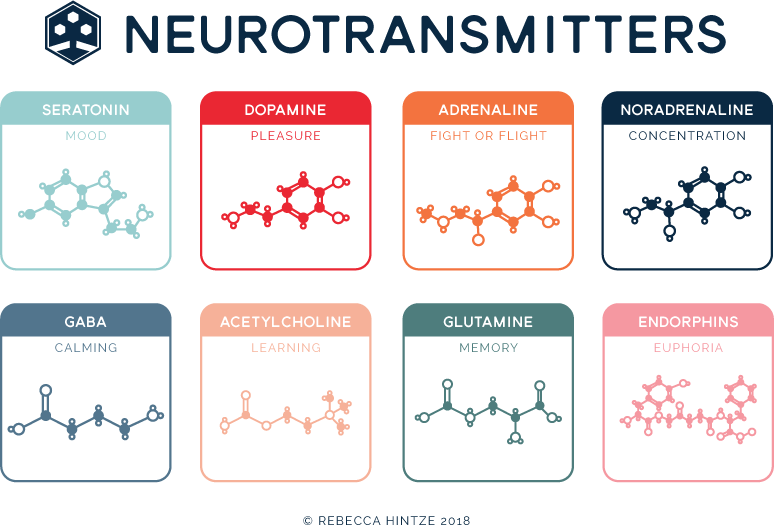 An increase in the concentration of serotonin in the synaptic cleft allows it to exercise its effects on postsynaptic receptors, what is the antidepressant effect of this group of drugs. Time required to decrease the activity of somatodendritic 5-HT autoreceptors 1A and the resulting release of serotonin from the axonal terminal explains the 2–3 week delay in the onset of the effect of SSRIs [49–51]. The main advantages of this group of drugs include their selective effect on the serotonin system, and the absence or minimal effect on other mediator systems of the brain, which minimizes side effects [3]. The selectivity of drugs in the SSRI group is not the same. As selectivity decreases, SSRIs can be ranked as follows: escitalopram, citalopram, sertraline, fluoxetine, paroxetine.
An increase in the concentration of serotonin in the synaptic cleft allows it to exercise its effects on postsynaptic receptors, what is the antidepressant effect of this group of drugs. Time required to decrease the activity of somatodendritic 5-HT autoreceptors 1A and the resulting release of serotonin from the axonal terminal explains the 2–3 week delay in the onset of the effect of SSRIs [49–51]. The main advantages of this group of drugs include their selective effect on the serotonin system, and the absence or minimal effect on other mediator systems of the brain, which minimizes side effects [3]. The selectivity of drugs in the SSRI group is not the same. As selectivity decreases, SSRIs can be ranked as follows: escitalopram, citalopram, sertraline, fluoxetine, paroxetine.
SNRIs (venlafaxine, milnacipran, duloxetine) inhibit serotonin reuptake along with inhibition of norepinephrine reuptake. The significance of norepinephrine disorders in depression will be discussed in future publications.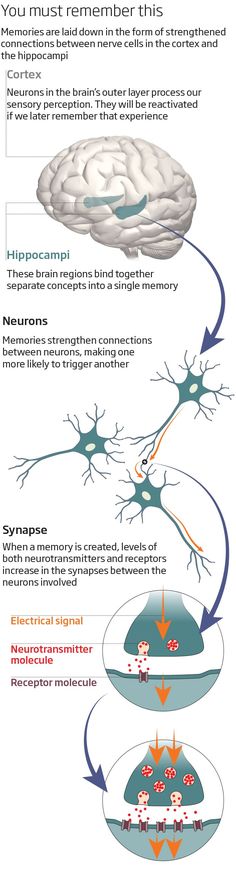 Blockade of serotonin reuptake is one of the main mechanisms of action of most TCAs (clomipramine, amitriptyline, doxepin, imipramine, protriptyline).
Blockade of serotonin reuptake is one of the main mechanisms of action of most TCAs (clomipramine, amitriptyline, doxepin, imipramine, protriptyline).
Unfortunately, the interaction of these drugs with other receptor systems (especially with cholinergic and histamine ones) leads to a large number of side effects and the refusal to use TCAs as first-line antidepressants [3].
Several drugs are active against 5-HT 1A receptors. Pindolol blocks presynaptic 5-HT 1A receptors and, therefore, should prevent an undesirable feedback effect, which is expressed in an increase in the concentration of somatodendritic serotonin. He showed the possibility of accelerating the onset of action of antidepressants [3]. Buspirone, gepirone, azaperone, partial antagonists of presynaptic 5-HT 1A receptors and activators of postsynaptic receptors have an antidepressant effect [3].
Antidepressants of various chemical groups have a blocking effect on 5-HT 2C receptors: tetracyclic (mianserin), noradrenergic and specific serotonergic (mirtazapine), serotonin modulators (nefazodone, trazodone), agonist M 1 - and M 2 melatonin receptors and 5-HT 2C receptor antagonist (agomelatine). The antidepressant activity of modern atypical antipsychotics is also associated with the blockade of 5-HT 2C - and 5-HT 2A receptors [3]. In addition to the antidepressant effect, these 5-HT 2 receptor antagonists synchronize biological rhythms disturbed during depression. In addition to the inhibition of 5-HT 2C receptors, mirtazapine, by blocking a2 receptors, stimulates the synthesis of serotonin [3].
The antidepressant activity of modern atypical antipsychotics is also associated with the blockade of 5-HT 2C - and 5-HT 2A receptors [3]. In addition to the antidepressant effect, these 5-HT 2 receptor antagonists synchronize biological rhythms disturbed during depression. In addition to the inhibition of 5-HT 2C receptors, mirtazapine, by blocking a2 receptors, stimulates the synthesis of serotonin [3].
Potentially interesting possibilities in the treatment of depression may be related to exposure to on 5-HT 1B/D -, 5-HT 6 - and 5-HT 7 - receptors. The emerging experimental data on the pharmacological efficacy of these targets require clinical validation [3].
Summarizing the presented data, we are fully aware that only an attempt was made to integrate modern information about the neurobiology of the serotonin system of the brain and the pharmacotherapy of depression based on the correction of serotonin metabolism disorders.![]() The results of many studies are beyond the scope of this review. The prism through which the selection of data for inclusion in the work was carried out was the possibility of practical refraction of the knowledge gained. After all, "there is nothing more practical than a good theory." Isolation of isolated serotonin dysfunction in depression is also very conditional. It is obvious that the activity of this neurotransmitter system must be considered in the structure of the complex of interrelationships of disorders of the noradrenergic, dopamine, GABA, peptidergic, and other mediator systems. The presented information, which is part of the modern molecular hypothesis of depression, must be supplemented with data on other biological disorders that occur in this disease. They will be reflected in our subsequent publications. We very much hope that the proposed information on the neurobiological mechanisms of depressive disorders will be useful to practitioners.
The results of many studies are beyond the scope of this review. The prism through which the selection of data for inclusion in the work was carried out was the possibility of practical refraction of the knowledge gained. After all, "there is nothing more practical than a good theory." Isolation of isolated serotonin dysfunction in depression is also very conditional. It is obvious that the activity of this neurotransmitter system must be considered in the structure of the complex of interrelationships of disorders of the noradrenergic, dopamine, GABA, peptidergic, and other mediator systems. The presented information, which is part of the modern molecular hypothesis of depression, must be supplemented with data on other biological disorders that occur in this disease. They will be reflected in our subsequent publications. We very much hope that the proposed information on the neurobiological mechanisms of depressive disorders will be useful to practitioners.
Literature
1. Kessler R.S. et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States: results from the National Comorbidity Survey // Arch Gen Psychiatry. - 1994. - Vol. 51. – P. 8-19.
Kessler R.S. et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States: results from the National Comorbidity Survey // Arch Gen Psychiatry. - 1994. - Vol. 51. – P. 8-19.
2. Murray C.J.L., Lopez A.D. Global burden of disease: a comprehensive assessment of mortality and morbidity from diseases, injuries and risk factors in 1990 and projects to 2020, Vol. I. - Harvard: World Health Organization, 1996.
3. Reasonable use of antidepressants: a technical review of the data prepared by the CINP Working Group / Ed. T. Bagai, H. Grunze, N. Sartorius: per. from English. - St. Petersburg, 2006. - 174 p.
4. Stein D.J. Serotonergic neurocircuitry in mood and anxiety disorders // Martin Dunitz Ltd. - 2003. - 82 p.
5. Mineka S., Watson D., Clark L.A. Comorbidity of anxiety and unipolar mood disorders // Annu Rev Psychol. - 1998. - Vol. 49.- P. 377-412.
6. MacLeod A.K., Byrne A. Anxiety, depression, and the anticipation of future positive and negative experience // J Abnorm Psychol. - 1993. - Vol. 102. - P. 238-247.
- 1993. - Vol. 102. - P. 238-247.
7. Damasio A.R. The somatic marker hypothesis and the possible function of the prefrontal cortex // Philos Trans R Sos. - 1996.- Vol. 54S. - P. 1413-1420.
8. MacLean P.D. Psychosomatic disease and the visceral brain: recent developments bearing on the Papez theory of emotion // Psychosom Med. - 1949. - Vol. 11. - P. 338-353.
9. Rolls E.T. A theory of emotion, and its application to understanding the neural basis of emotions // Cognition Emotion. - 1990. - Vol. 4. - P.161-190.
10. Videbach P. PET measurements of brain glucose metabolism and blood flow in major depression: a critical review // Acta Psychiatr Scand. - 2000. - Vol. 101. - P. 11-20.
11. Narushima K., Kosier J.T., Robinson R.G. A reappraisal of poststroke depression, intra- and inter-hemispheric lesion location using meta-analysis // J Neuropsychiatry Clin Neurosci. - 2003. - Vol. 15. - P. 422-430.
12. Shimoda K., Robinson R.G. The relationship between poststroke depression and lesion location in long-term follow-up // Biol Psychiatry. - 1999. - Vol. 45. – P. 187-192.
- 1999. - Vol. 45. – P. 187-192.
13. Camus V., Kraehenbuhl H., Preisig M. et al. Geriatric depression and vascular diseases: what are the links? // J Affect Discord. - 2004. - Vol. 81, No. 1. - P. 1-16.
14. Firbank M.J., Lloyd A.J., Ferrier N., O'Brien J.T. A volumetric study of MRI signal hyperintensities in late-life depression // Am J Geriatr Psychiatry. - 2004. - Vol. 12, No. 6. - P. 606-612.
15. Seki T., Awata S., Koizumi Y. et al. Association between depressive symptoms and cerebrovascular lesions on MRI in community-dwelling elderly individuals // Nippon Ronen Igakkai Zasshi. - 2006. - Vol. 43, N 1. - P. 102-107.
16. Dahlstrom A., Fuxe K. Evidence for the existence of monoamine neurons in the central nervous system // Acta Physiol Scand. - 1965.-Vol. 64. – P. 1-85.
17. Barkhatova V.P. Neurotransmitters and extrapyramidal pathology. - M.: Medicine, 1988.
18. Gromova E.A. Serotonin and its role in the body. - M.: Medicine, 1966.
19. Lutsenko N.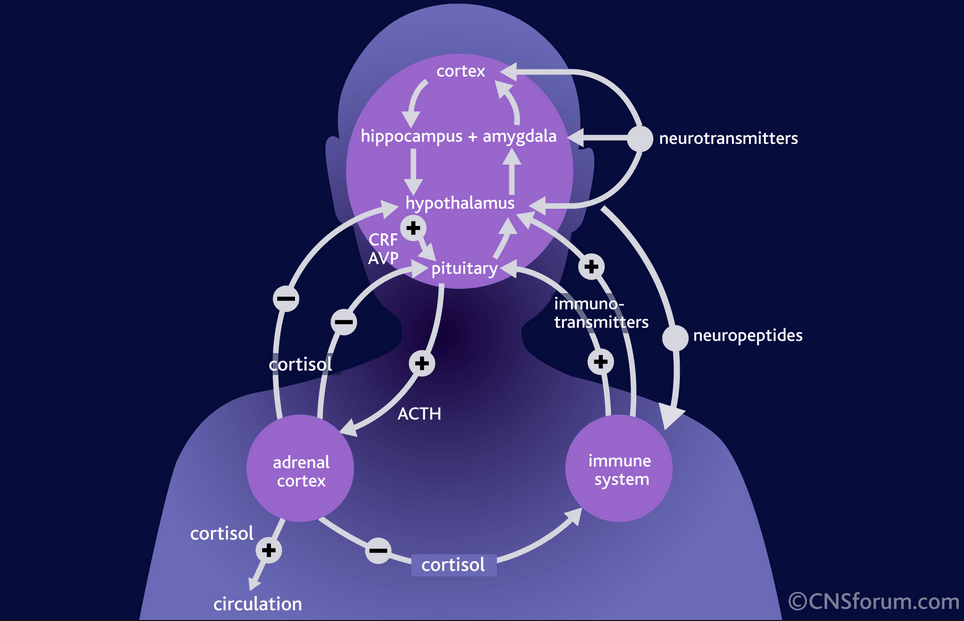 G., Suvorov N.N. Regulation of serotonin biosynthesis in the central nervous system // Uspekhi sovrem. biol. - 1982. - T. 94. - S. 243-251.
G., Suvorov N.N. Regulation of serotonin biosynthesis in the central nervous system // Uspekhi sovrem. biol. - 1982. - T. 94. - S. 243-251.
20. Bremmer J.D., Innis R.B., Salomon R.M. et al. Positron emission tomography measurement of cerebral metabolic correlates of tryptophan depletion-induced depressive relapse // Arch Gen Psychiatry. - 1997.-Vol. 54. – P. 364-374.
21. Konysova A.Zh. Serotonin metabolism in multiple sclerosis and retrobulbar neuritis (clinical and biochemical study): Diss. …cand. honey. Sciences. M., 1995.
22. Sergeev P.V. Receptors. – Volgograd, 1999.
23. Cox C., Cohen J. 5-HT2B receptor signaling in the rat stomach fundus: dependence on calcium influx, calcium release and protein kinase C // Behav. Brain Res. - 1996. - Vol. 73. – P. 289.
24. Fox S.H., Brotchie J.M. Anti-parkinsonian action of 5-HT2C receptor antagonism in the substantia nigra pars reticulata // Mov. Discord. - 1997. - Vol. 12, Suppl. 1. – P. 116.
25. Hanssen E., Nilsson A. , Ericsson P. Heterogeneity among astrocytes evaluated biochemical parameters // Adv. biosci. - 1986. -
, Ericsson P. Heterogeneity among astrocytes evaluated biochemical parameters // Adv. biosci. - 1986. -
Vol. 61. - P. 235-241.
26. Holstege J.S., Knypers H.G. Brainstem projections to spinal motoneurons: an update commentary // Neuro. sci. - 1987. - Vol. 23. – P. 809-821.
27. Blier P., Ward N.M. Is the role for 5HT-1A-agonists in the treatment of depression // Biol. Psychiatry. - 2003. - Vol. 53. – P. 193-203.
28. Connor J.D. et al. Use of GR 55562, a selective 5-HT1D antagonist, to investigate 5-HT1D receptor subtypes mediating cerebral vasoconstriction // Cephalgia. - 1995. - Vol. 15, Suppl. 14. – P. 99.
29. Choi C, Maroteaux J. Immunohistochemical localization of the serotonin 5-HT2B receptor in mouse gut, cardiovascular system, and brain // FEBS Lett. - 1996. - Vol. 391. – P. 45.
30. Martin G.R. et al. 5-HT2C receptor agonists and antagonists in animal models of anxiety // Eur. Neuropharmacol. - 1995.-Vol. 5. – P. 209.
31. Misyuk N.S. et al. Materials for the exchange of serotonin in inhibitory states of the brain. – Minsk, 1965.
Materials for the exchange of serotonin in inhibitory states of the brain. – Minsk, 1965.
32. Willner P. Validity, reliability and utility of chronic mild stress model of depression: a 10 years review and evaluation // Psychopharmacology. - 1997. - Vol. 134. - P. 319-329.
33. Papp M., Cruca P., Boyer P.-A., Mocaer E. Effect of agomelatine in the chronic mild stress model of depression in the rat // Neuropsychopharmacology. - 2003. - Vol. 28. – P. 694-703.
34. Golubev V.L., Levin Ya.I., Vein A.M. Parkinson's disease and parkinsonism syndrome. - M.: MEDpress, 1999.
A complete list of references, including 51 items, is in the editorial office.
bases of the neurocirculatory and neurotrophic hypothesis of depression
Depression is one of the most frequent mental illnesses faced by doctors of various profiles. This also applies to major depressive disorder (MDD). According to the WHO, its relative prevalence during life is 16.2%, during the year - 6. 6% [27].
6% [27].
The diagnosis of MDD according to the criteria of the American classification DSM-IV [10] [1] , designated major depressive disorder (MDD), requires the presence of at least 5 symptoms for at least 2 weeks in the development of major depressive episodes (MDE): low mood, feelings of worthlessness/guilt, thoughts of death or suicidality, loss of interest in the environment, loss of appetite, sleep disturbances, psychomotor changes, hypo- or anergy, disturbances in concentration and indecision.
MDD as a progressive disease
Although MDD is often characterized as an episodic disease, prospective studies in recent years have shown that it tends to be more recurrent. Thus, a 15-year study [51] of 380 patients showed that 73% of patients with BDE had a recurrent course of the disease. At the same time, the development of each BDE increased the risk of developing the next one. Similar results were obtained in the framework of the international STARD (Sequenced Treatment Alternatives to Relieve Depression) project [17], in which 1500 patients with MDD were observed, of which 74% had more than one PDE.
It is assumed that the recurrent course of MDD is triggered by the development of a number of neurobiological disorders that increase the body's vulnerability to the development of a new BDE. In accordance with this, the “kindling hypothesis” was put forward [38], according to which each PDE becomes a trigger for the onset of a new depressive episode and this phenomenon intensifies over time. As the number of BDEs increases, their development later on becomes more and more related to their previous number, and not to stress factors in the patient's life [26]. S. Monroe and K. Harkness [38] explain the swaying of the disease with an increase in the duration of the disease by a decrease in the number of depressive episodes of the protective threshold (sensitization of the body to stress) and/or an increase in spontaneous dysregulation of the neurobiological systems responsible for the development of MDD. K. Kendler et al. [26], when analyzing the risk of developing recurrent seizures in a cohort of twins, showed a large contribution of the genetic factor to the development of the “rocking” phenomenon.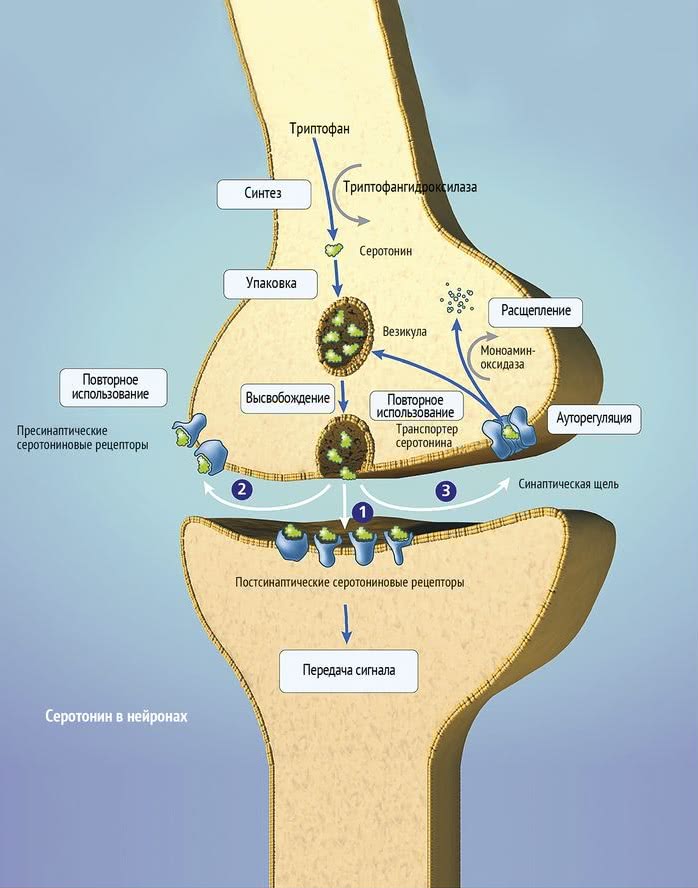
This study also found a reduction in the magnitude of the association between stressors and the onset of recurrent MDD in patients at high genetic risk for MDD.
Risk factors for the development of a recurrent course of MDD are also family history of depression, early separation of the child from the mother (deprivation), and inadequate treatment at the initial stages of the disease [17, 23, 28]. Some researchers [16] believe that any aggravating events in a patient's life from birth to the onset of the disease increase the risk of recurrent attacks of MDD.
The chronization of the course of MDD suggests the presence of slowly increasing neurobiological consequences, leading to the "rocking" of the disease. This makes the practical recovery of patients with MDD less and less likely, especially in the late stages of the disease. If for patients with a disease duration of less than 1 year, the probability of recovery is 16%, then for patients suffering from MDD for more than 5 years, it is less than 1% [24]. The longer the period observed between previous and subsequent PDEs, the higher the likelihood of recovery. If it exceeds 1 year in the early stages of the disease, then the probability of not resuming a new episode reaches 20% [52].
The longer the period observed between previous and subsequent PDEs, the higher the likelihood of recovery. If it exceeds 1 year in the early stages of the disease, then the probability of not resuming a new episode reaches 20% [52].
The recurrence and chronicity of MDD reduce the prospects for treating the disease, while its goal is not the reduction or complete absence of all the main symptoms of the disease, but the lengthening of the periods between attacks [25].
Functional and structural brain changes in MDD
A number of neurobiological abnormalities associated with MDD have now been identified. They are especially pronounced in the limbic structures and their connections, which are related to the regulation of affect. These neuroanatomical zones include the medial, orbitofrontal, and dorsolateral prefrontal cortex, the anterior cingulate cortex, the ventral striatum, which includes the nucleus accumbens, the amygdala, and the hippocampus.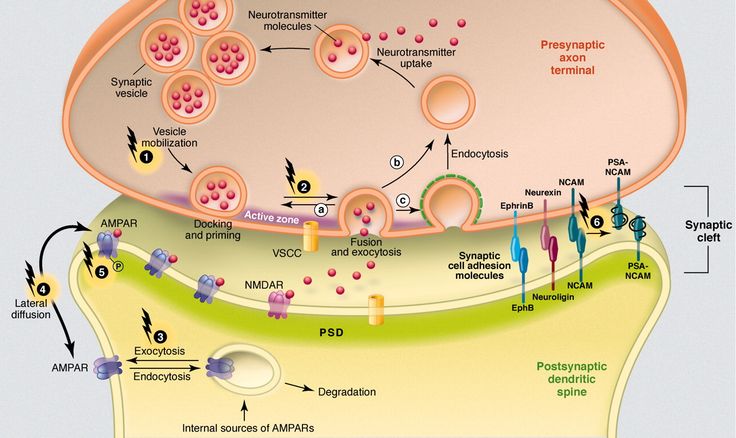 It is assumed [8] that the anomalies found in these structures in patients with MDD form the basis for its formation.
It is assumed [8] that the anomalies found in these structures in patients with MDD form the basis for its formation.
As an integrative circuit, the prefrontal cortex, cingulate cortex, amygdala, and hippocampus provide not only mood regulation, but also learning and contextual memory. In addition, relevant areas of the prefrontal cortex are related to pain, aggression, sexual functioning and eating behavior, development of affective disorders, support of executive functions, attention and working memory [55]. Separate parts within the anterior cingulate cortex have different functions: the dorsal part of the anterior cingulate cortex provides some cognitive and executive functions, the ventral part of the anterior cingulate cortex processes emotional and motivational information; anterior cingulate cortex — monitoring of behavior and cognitive functions [6, 35].
In patients with MDD, hyperactivation of the ventromedial and orbitofrontal zones of the prefrontal cortex was found, as well as a decrease in the activity of the dorsolateral prefrontal cortex [11].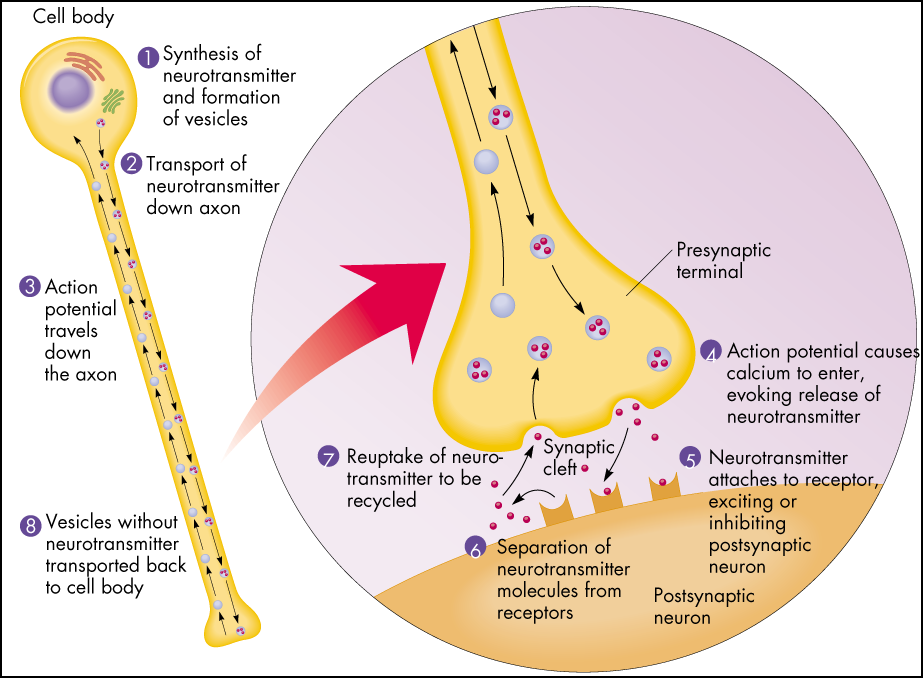 Based on the functions of these brain regions, it was suggested that these disorders are responsible for the manifestation of symptoms associated with MDD. Hyperactivity of the ventromedial, prefrontal cortex is associated with increased sensitivity to pain, increased anxiety, depressive rumination, and tension. Hypoactivity of the dorsolateral prefrontal cortex can cause psychomotor retardation, apathy, and deficits in attention and working memory. When using the method of magnetic resonance imaging (MRI), a decrease in “communicativeness” between the amygdala and the anterior cingulate cortex was found [1]. Due to the loss of these connections, the anterior cingulate cortex loses its inhibitory ability to act as an emotional regulator, which leads to the development of a motivational and affective gap [62].
Based on the functions of these brain regions, it was suggested that these disorders are responsible for the manifestation of symptoms associated with MDD. Hyperactivity of the ventromedial, prefrontal cortex is associated with increased sensitivity to pain, increased anxiety, depressive rumination, and tension. Hypoactivity of the dorsolateral prefrontal cortex can cause psychomotor retardation, apathy, and deficits in attention and working memory. When using the method of magnetic resonance imaging (MRI), a decrease in “communicativeness” between the amygdala and the anterior cingulate cortex was found [1]. Due to the loss of these connections, the anterior cingulate cortex loses its inhibitory ability to act as an emotional regulator, which leads to the development of a motivational and affective gap [62].
Being at the intersection of limbic, cognitive, executive and neuroendocrine regulatory pathways, including the hypothalamic-pituitary-adrenal axis, the hippocampus should be particularly vulnerable to MDD.![]() P. Videbech and B. Ravnkilde [60], summarizing 12 studies using a meta-analysis, found that the volume of the hippocampus was significantly reduced in MDD compared with the norm, this decrease was observed bilaterally with a slight prevalence on the right side. A number of other studies [8, 41] have shown that the decrease in hippocampal volume is directly proportional to the number of BDEs and the duration of MDD. Even in the period of remission after BDE, patients continue to experience a slow decline in the already reduced volume of the hippocampus [7].
P. Videbech and B. Ravnkilde [60], summarizing 12 studies using a meta-analysis, found that the volume of the hippocampus was significantly reduced in MDD compared with the norm, this decrease was observed bilaterally with a slight prevalence on the right side. A number of other studies [8, 41] have shown that the decrease in hippocampal volume is directly proportional to the number of BDEs and the duration of MDD. Even in the period of remission after BDE, patients continue to experience a slow decline in the already reduced volume of the hippocampus [7].
The difference in hippocampal volume between patients with MDD and controls is not always related to the disease. Studies [32, 54] showed that the genetic contribution to the reduced volume of the hippocampus is 54%. Studies of autopsy brain tissue [53] of patients with MDD showed a decrease in the size of the hippocampus due to an increase in the density of neurons and a decrease in the volume of the neuropil (due to a decrease in dendritic branching).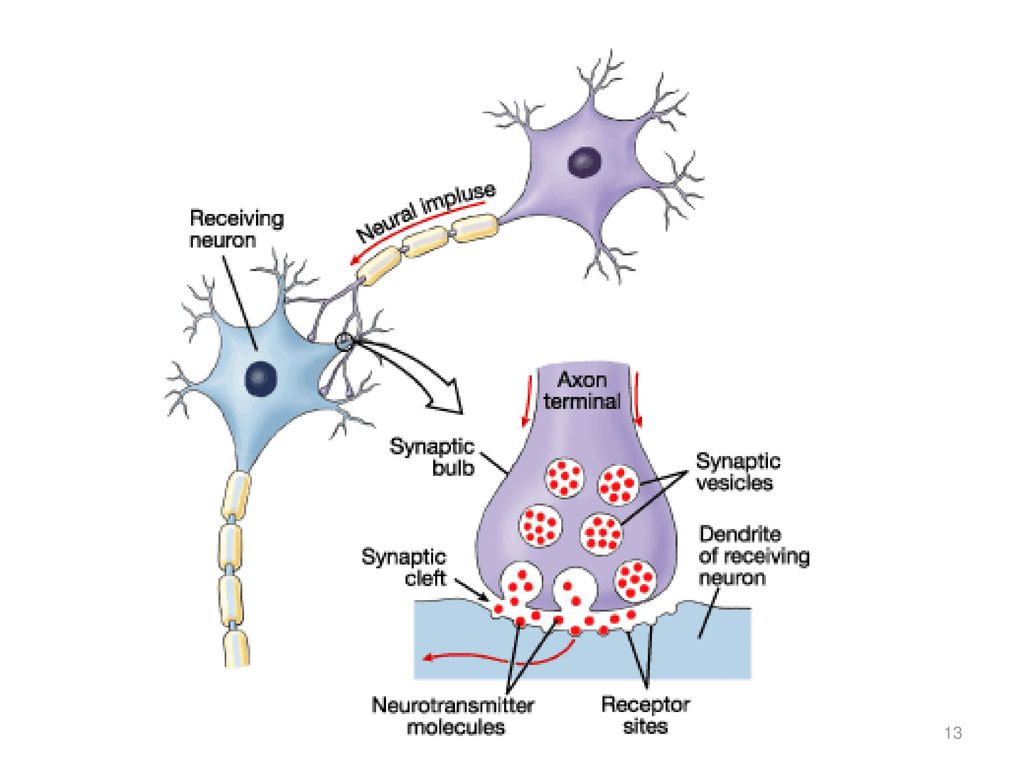
The pooled results of genetic, neuroanatomical, and clinical studies suggest that a reduction in the size of the hippocampus is a predisposing factor for MDD. During treatment, the volume of the hippocampus is not normalized, which leads to the impossibility of a complete recovery of the patient.
Molecular processes mediating neurobiological changes in MDD
Violations in the hippocampus by the feedback mechanism can lead to dysregulation of the function of the structures concerned. In this case, a high level of the main stress hormone cortisol, which affects neuroplasticity and cell resistance, plays an important role [33]. The disturbed balance between the hormones of the glucocorticoid and mineralocorticoid groups, as well as the high density of glucocorticoid receptors (GR) in the brain in MDD, lead to the vulnerability of hippocampal neurons [9]. As a result, atrophy of hippocampal cells develops, which leads to even greater neuroendocrine dysfunction and loss of controllability of the hippocampus-mediated brain systems [40].![]() As a result of an increased level of glucocorticoids (in particular, cortisol) and a decrease in the functional activity of the hippocampus, a decrease in the sensitivity of GH is observed. Under conditions of chronic stress (with elevated levels of cortisol), a decrease in GH sensitivity can have negative consequences, since insufficient signaling through GR “turns off” the feedback mechanism of protection against stress [46]. As a result, hypothalamic-pituitary-adrenal hyperactivation occurs, which, combined with the activation of the function of the amygdala of the brain, increases sympathetic tone, which in turn causes an increased release of pro-inflammatory cytokines from macrophages. Increased secretion of pro-inflammatory cytokines (interleukins 1 and 6 and tumor necrosis factor, TNF-α) reduces insulin levels and GH sensitivity, which aggravates metabolic and neuroendocrine disorders [59]. Clinically, such disorders are manifested by symptoms of fatigue, loss of appetite and libido, as well as hypersensitivity to pain [59].
As a result of an increased level of glucocorticoids (in particular, cortisol) and a decrease in the functional activity of the hippocampus, a decrease in the sensitivity of GH is observed. Under conditions of chronic stress (with elevated levels of cortisol), a decrease in GH sensitivity can have negative consequences, since insufficient signaling through GR “turns off” the feedback mechanism of protection against stress [46]. As a result, hypothalamic-pituitary-adrenal hyperactivation occurs, which, combined with the activation of the function of the amygdala of the brain, increases sympathetic tone, which in turn causes an increased release of pro-inflammatory cytokines from macrophages. Increased secretion of pro-inflammatory cytokines (interleukins 1 and 6 and tumor necrosis factor, TNF-α) reduces insulin levels and GH sensitivity, which aggravates metabolic and neuroendocrine disorders [59]. Clinically, such disorders are manifested by symptoms of fatigue, loss of appetite and libido, as well as hypersensitivity to pain [59].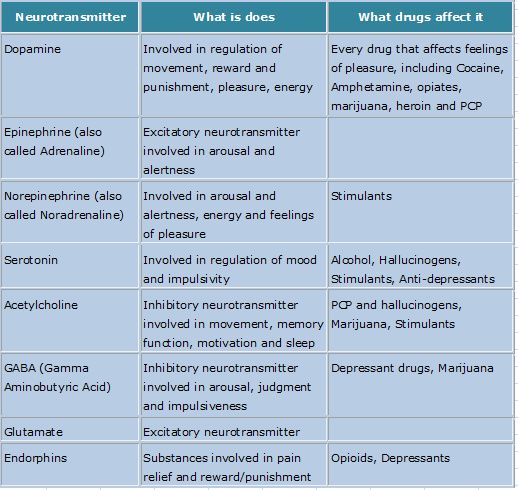
Pro-inflammatory cytokines can also reduce neurotrophic cell support and monoamine neurotransmission, which in turn leads to neuronal apoptosis and impaired glial function and neuron-glial relationships in MDD [47, 61]. The significance of such relationships is determined by the fact that glial cells are involved in complex interactions with neurons, maintain the homeostasis of the neuron and its environment by regulating the content of electrolytes, neurotransmitters, cytokines, and neurotrophic factors [3]. Recently, much attention has been paid not only to astroglia, but also to microglia, which is associated with immune dysregulation and additional production of the release of pro-inflammatory cytokines [19]. Neurons, in turn, reciprocally support the functioning of glia through the production of neurotrophin.
Brain-derived neurotrophic factor (BDNF) plays an integral role in maintaining normal neuron-glial interaction [12]. Being involved in neurogenesis, BDNF is the main neurotrophin in the hippocampus.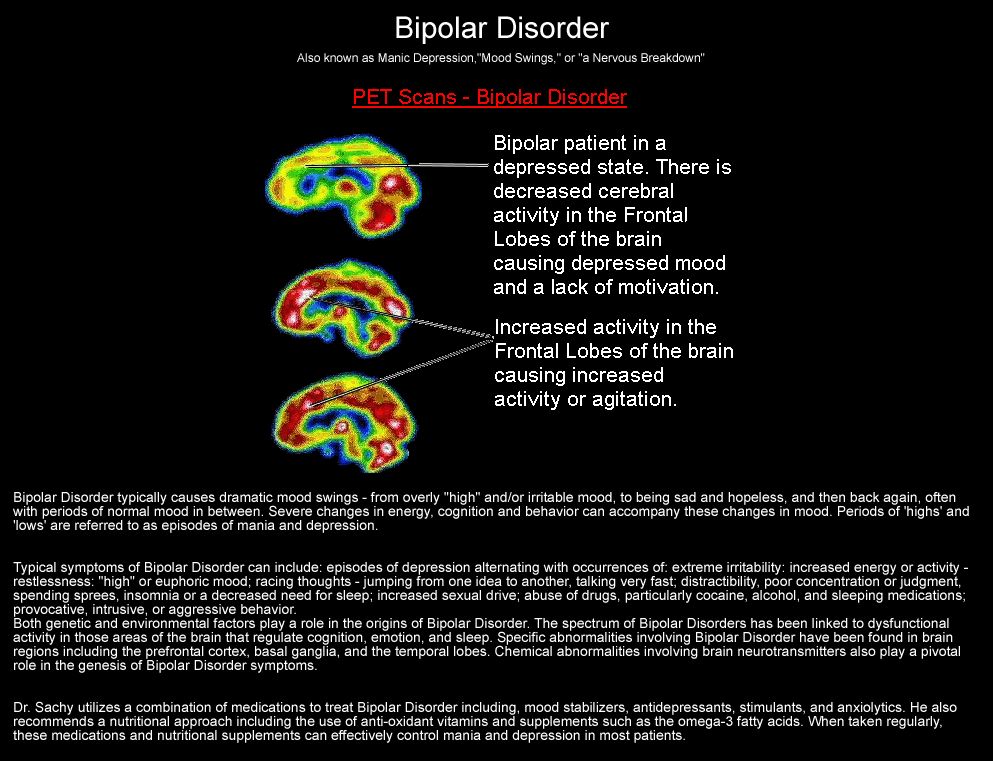 As a dimeric protein involved in cell maintenance, plasticity, growth, and death (apoptosis), BDNF is structurally related to nerve growth factor and is widely distributed in the brain [57]. BDNF, interacting with tyrosine kinase receptors, causes cellular resistance to external factors and long-term potentiation effects. However, the precursor of BDNF, pro-BDBF, by binding to the p75 receptor, can cause a forced reduction of such structures providing intercellular contacts as dendritic spines and cell death. Depending on the amount of BDNF expression, this process can be expressed to varying degrees. This process is regulated by various neurotransmitters (glutamate, gamma-aminobutyric acid, serotonin, norepinephrine, acetylcholine, dopamine, and hormones) [31].
As a dimeric protein involved in cell maintenance, plasticity, growth, and death (apoptosis), BDNF is structurally related to nerve growth factor and is widely distributed in the brain [57]. BDNF, interacting with tyrosine kinase receptors, causes cellular resistance to external factors and long-term potentiation effects. However, the precursor of BDNF, pro-BDBF, by binding to the p75 receptor, can cause a forced reduction of such structures providing intercellular contacts as dendritic spines and cell death. Depending on the amount of BDNF expression, this process can be expressed to varying degrees. This process is regulated by various neurotransmitters (glutamate, gamma-aminobutyric acid, serotonin, norepinephrine, acetylcholine, dopamine, and hormones) [31].
Preclinical and clinical studies indicate that BDNF dysregulation occurs in chronic stress and depression. In animal models of depression, a decrease in the expression of BDNF synthesis in brain tissues was shown [15] (similar results were obtained using models with acute or chronic pain stimulation in animals). Decreased serum BDNF levels have been found in untreated patients with MDD compared with treated patients or healthy controls [50]. Similar results were obtained on autopsy material from the brain of patients with MDD (after suicide): a decrease in the level of BDNF and neurotrophin type NT-3 compared with the material obtained from patients with MDD who did not die due to suicide) [22].
Decreased serum BDNF levels have been found in untreated patients with MDD compared with treated patients or healthy controls [50]. Similar results were obtained on autopsy material from the brain of patients with MDD (after suicide): a decrease in the level of BDNF and neurotrophin type NT-3 compared with the material obtained from patients with MDD who did not die due to suicide) [22].
The above data allowed the famous Venezuelan scientist Fuad Lechin to formulate the so-called neurocirculatory and neurotrophic hypothesis of MDD pathogenesis [29]. This hypothesis is based on the idea of the existence of links between systems of neurotransmitters or neurotrophic factors in the periphery (blood, cerebrospinal fluid, etc.) and systems of relationships between the same neurotransmitters or neurotrophic factors in the CNS in depression (MDD) and other mental disorders. As part of the development of this hypothesis, it was shown that stress and genetic vulnerability, through changes in the system of relationships between mediators, neurohumoral and neurotrophic factors in the periphery, increase the production of central glucocorticoid steroids, which leads to impaired cell plasticity and a decrease in the system of growth factors and GR sensitivity [14].![]() At the same time, there is also a decrease in the synthesis of neurotrophic growth factors, such as BDNF (in the blood and CNS). This causes negative structural and functional changes in the limbic system, especially in the hippocampus. In chronic and recurrent forms of MDD, gradual atrophy of the hippocampus occurs, followed by dysregulation in the neurocircuits of the limbic system [48]. According to this hypothesis, the recovery or remission of MDD depends on the reversibility of these processes, especially on the increase in BDNF synthesis during treatment.
At the same time, there is also a decrease in the synthesis of neurotrophic growth factors, such as BDNF (in the blood and CNS). This causes negative structural and functional changes in the limbic system, especially in the hippocampus. In chronic and recurrent forms of MDD, gradual atrophy of the hippocampus occurs, followed by dysregulation in the neurocircuits of the limbic system [48]. According to this hypothesis, the recovery or remission of MDD depends on the reversibility of these processes, especially on the increase in BDNF synthesis during treatment.
Within the framework of the neurocirculatory and neurotrophic BDD hypothesis, monoamine theories (serotonin, norepinephrine) become complementary. Recall that, according to monoamine theories, depression is associated with low levels of monoamine neurotransmitters, especially serotonin and norepinephrine. Recent MRI studies of patients with untreated depression have shown an increase in the protein density of the enzyme monoamine oxidase A (MAO-A), which has a non-specific enzymatic activity for both serotonin and norepinephrine. Therefore, the modern version of the monoamine theory of depression postulates that a long-term decrease in the levels of serotonin and norepinephrine due to the activation of MAO-A in various parts of the brain of patients leads to a disruption in the functioning of serotonin transporters (SERT) and noradrenaline, and as a result, to an exacerbation of depression [37]. Serotonin- and noradrenergic ascending nerve fibers originate from the nuclei of the brainstem and innervate the limbic system, the prefrontal cortex associated with structures involved in mood regulation; descending pathways pass through the dorsolateral parts of the spinal cord and are associated with the regulation of the pain threshold [36]. Therefore, depending on the magnitude of changes in the functional activity of SERT or the norepinephrine transporter protein, various clinical manifestations of depression may be observed in the corresponding regions of the brain [37].
Therefore, the modern version of the monoamine theory of depression postulates that a long-term decrease in the levels of serotonin and norepinephrine due to the activation of MAO-A in various parts of the brain of patients leads to a disruption in the functioning of serotonin transporters (SERT) and noradrenaline, and as a result, to an exacerbation of depression [37]. Serotonin- and noradrenergic ascending nerve fibers originate from the nuclei of the brainstem and innervate the limbic system, the prefrontal cortex associated with structures involved in mood regulation; descending pathways pass through the dorsolateral parts of the spinal cord and are associated with the regulation of the pain threshold [36]. Therefore, depending on the magnitude of changes in the functional activity of SERT or the norepinephrine transporter protein, various clinical manifestations of depression may be observed in the corresponding regions of the brain [37].
The role of neurotransmitters in the development of remission in MDD
A large number of studies have shown that the treatment of MDD with selective serotonin reuptake blockers (SSRIs) or norepinephrine (SNRIs) in the brain of patients increases the levels of serotonin or noradrenaline, respectively.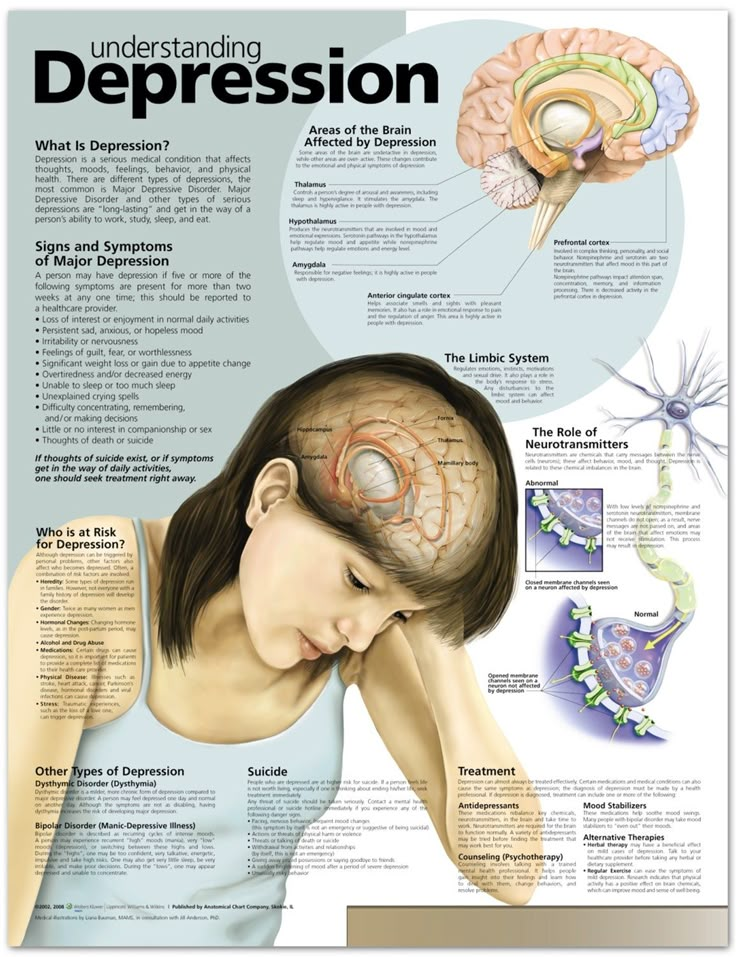 Long-term treatment with these drugs also increases the level of cyclic adenosine monophosphate (cAMP) in the brain, which stimulates specific protein kinase A. Activation of this enzyme activates the state of the part of the cell genome responsible for the synthesis of BDNF [12]. Antidepressant-induced cAMP production also increases GR sensitivity, inhibits the negative effect of increased production of cytokines, and thereby restores their functional activity and regulation in the corresponding neurocircuits [43].
Long-term treatment with these drugs also increases the level of cyclic adenosine monophosphate (cAMP) in the brain, which stimulates specific protein kinase A. Activation of this enzyme activates the state of the part of the cell genome responsible for the synthesis of BDNF [12]. Antidepressant-induced cAMP production also increases GR sensitivity, inhibits the negative effect of increased production of cytokines, and thereby restores their functional activity and regulation in the corresponding neurocircuits [43].
The effect of increasing levels of monoamines (dopamine, serotonin and norepinephrine) during treatment with antidepressants, associated with an increase in the synthesis of BDNF and other neurotrophic factors, is one of the leading ones in the mechanism of their antidepressant action. Animal studies [21] have shown that an increase in the levels of monoamines (serotonin and norepinephrine) during chronic administration of antidepressants causes an increase in the level of BDNF in brain astrocytes. In clinical studies, it has been shown that when patients with MDD are successfully treated with antidepressants, the serum level of BDNF is normalized. It is believed that the serum level of BDNF reflects its synthesis in the brain. This is supported by animal experiments, as well as by the fact that BDNF freely passes through the blood-brain barrier [30]. It has been shown that the degree of improvement in the condition of patients treated with antidepressants with simultaneous inhibition of serotonin and norepinephrine reuptake (dual reuptake inhibitors) significantly correlates with an increase in the level of serum BDNF [2]. Studies of post-mortem brain samples showed that in cases of death of patients during treatment with dual reuptake inhibitors, a higher content of BDNF was determined in its tissue than in brain samples taken from untreated patients [49].
In clinical studies, it has been shown that when patients with MDD are successfully treated with antidepressants, the serum level of BDNF is normalized. It is believed that the serum level of BDNF reflects its synthesis in the brain. This is supported by animal experiments, as well as by the fact that BDNF freely passes through the blood-brain barrier [30]. It has been shown that the degree of improvement in the condition of patients treated with antidepressants with simultaneous inhibition of serotonin and norepinephrine reuptake (dual reuptake inhibitors) significantly correlates with an increase in the level of serum BDNF [2]. Studies of post-mortem brain samples showed that in cases of death of patients during treatment with dual reuptake inhibitors, a higher content of BDNF was determined in its tissue than in brain samples taken from untreated patients [49].
One study [34] noted that a positive response to antidepressant treatment was accompanied by normalization of cortical activity.![]() Already after 1 week of treatment, the activity of the hippocampus increased and the activity of the posterior cingulate and prefrontal cortex (posterior cingulate and prefrontal cortex) decreased in patients. After 6 weeks of treatment, responder patients showed signs of normalization of limbic system activity and an increase in prefrontal cortex activity, while there were no changes in resistant patients after the first week. It also turned out that the normalization of the functioning of the amygdala and the anterior cingulate cortex is associated with a positive response to treatment. In addition, antidepressant-resistant patients with MDD have been found to have elevated levels of pro-inflammatory cytokines compared with control or euthymic MDD patients [42].
Already after 1 week of treatment, the activity of the hippocampus increased and the activity of the posterior cingulate and prefrontal cortex (posterior cingulate and prefrontal cortex) decreased in patients. After 6 weeks of treatment, responder patients showed signs of normalization of limbic system activity and an increase in prefrontal cortex activity, while there were no changes in resistant patients after the first week. It also turned out that the normalization of the functioning of the amygdala and the anterior cingulate cortex is associated with a positive response to treatment. In addition, antidepressant-resistant patients with MDD have been found to have elevated levels of pro-inflammatory cytokines compared with control or euthymic MDD patients [42].
A study of the reduction of symptoms of MDD during treatment showed its connection with regional changes in the metabolism of the brain of patients [4]. Thus, a decrease in the severity of cognitive impairments correlated with an increase in the activity of the dorsal part of the anterior cingulate cortex, and a decrease in the manifestations of fatigue syndrome and psychomotor retardation was associated with a decrease in the activity of the ventromedial zone of the prefrontal cortex. Interestingly, these correlations were observed regardless of the conduct of psychopharmaco- or psychotherapy.
Interestingly, these correlations were observed regardless of the conduct of psychopharmaco- or psychotherapy.
There is a point of view [13] that the restoration of neurobiological regulation in MDD through an increase in the synthesis of neurotrophic factors and, as a result, neurogenesis, is probably the general radical that determines the effectiveness of treatment, regardless of whether it is psychopharmacological, psychotherapeutic or somatic (in in the latter case, they mean the use of a diet and special physical exercises).
The presence of the effect of "rocking" and chronification of MDD during its long course makes it urgent to find the most effective therapy already at the first attack of the disease. Long-term studies have shown that the best predictor of the course of the disease is the patient's response to treatment already at the first attack, i.e. during the first 6 weeks from the onset of the attack [56]. This is of particular importance for elderly patients, in whom previous inadequate antidepressant treatment may lead to drug resistance [58].![]() A positive patient response to antidepressant treatment at an early stage of the disease gives a chance that further improvement in the quality of psychopharmacotherapy will lead to a complete or partial recovery of the patient [5].
A positive patient response to antidepressant treatment at an early stage of the disease gives a chance that further improvement in the quality of psychopharmacotherapy will lead to a complete or partial recovery of the patient [5].
One of the ways to improve the quality of treatment of patients with MDD is the use of antidepressants with a broader spectrum of action or an appropriate complex of drugs with different mechanisms of action [39]. However, the results of a large meta-analysis [44], which included 92 studies (17,036 patients), showed the same efficacy of serotonin- and noradrenergic antidepressants. However, a new generation of antidepressants that are dual selective reuptake inhibitors (both serotonin and norepinephrine) - dual antidepressants have proven to be more effective than SSRIs and SNRIs. Dual antidepressants were especially effective if there were pain symptoms in the pattern of depression [18, 20], which is known to be characteristic of masked depression.![]() This is due to the fact that in the treatment of dual antidepressants there is a synergistic activation of the serotonin and norepinephrine systems.
This is due to the fact that in the treatment of dual antidepressants there is a synergistic activation of the serotonin and norepinephrine systems.
The neurocirculatory and neurotrophic hypotheses of depression show that the factors initiating an episode in MDD and the factors supporting the development of the disease are different. At the initial stage, vulnerability to stress, genetic predisposition to the disease interact with each other and initiate a cascade of neurobiological disorders that destroy normal dynamic connections in the brain areas associated with the regulation of mood, cognition, physical and mental activity, pain sensations, etc. In chronic disease as MDD progresses, further structural and functional disorders begin to potentiate.
The main goal of treatment in the presence of the consequences of chronic MDD is to restore normal functioning and prevent further structural and functional neurobiological disorders in the brain of patients. An increase in serotonin- and noradrenergic neurotransmission with the help of antidepressants initiates the restoration of the synthesis of neurotrophic factors (especially BDNF), which normalizes the activity of glucocorticoids and neuroendocrine regulation.![]() The use of dual antidepressants (serotonin and norepinephrine reuptake inhibitors) increases the likelihood of achieving remission due to the complex reduction of emotional and somatic symptoms (including pain) in depression.
The use of dual antidepressants (serotonin and norepinephrine reuptake inhibitors) increases the likelihood of achieving remission due to the complex reduction of emotional and somatic symptoms (including pain) in depression.
According to the neurocirculatory and neurotrophic hypothesis, the results of treatment in the early stages of the disease determine the prognosis and recurrence of the course of MDD. The presence of residual symptoms during treatment also affects the further course of the disease. When remission is achieved, patients should be informed of the greater benefit of continuing long-term, continuous treatment than episodic or incomplete treatment. Treatment, which includes individual or group cognitive therapy along with active psychopharmacotherapy, contributes to the correction of molecular factors in the pathogenesis of the disease, which reduces the subsequent risk of MDD exacerbation [45]. When remission is achieved, maintaining brain metabolism at the normal level is a more important task than preventing a developing exacerbation.