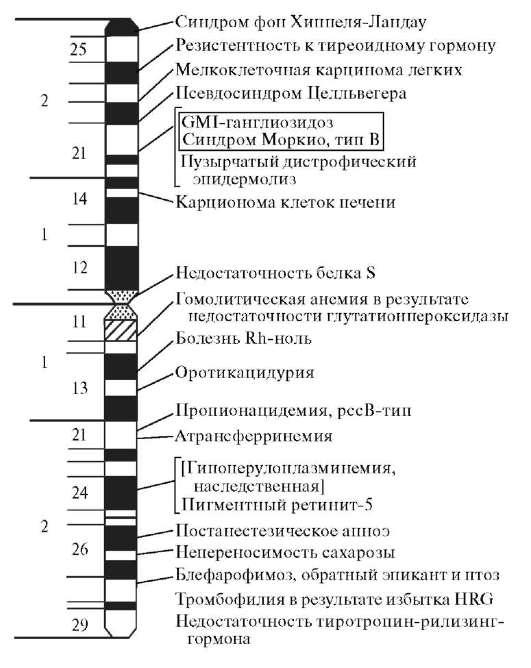
Chromosome 14 hoarding
The Genetics of Compulsive Hoarding
Is compulsive hoarding inherited?
People who compulsively acquire and hoard clutter to the extent that it impairs their daily activities are labeled “compulsive hoarders.” The condition is classed as a subtype of obsessive-compulsive disorder (OCD), present in 30 to 40 percent of individuals affected with OCD. It may damage relationships, cut the individual off from society, and even endanger lives.
Compulsive hoarding is distinct from bad planning and disorganization because it is believed to be a pathological brain disorder. It is often a symptom of other disorders, such as impulse control disorder or attention-deficit hyperactivity disorder. Bereavement or another significant life event can trigger excessive hoarding behavior.
Hoarding often runs in families, but it is uncertain whether DNA is involved. “People with this problem tend to have a first-degree relative who also does,” says Randy O. Frost, Ph.D., a psychologist at Smith College, Northampton, Massachusetts. “So it might be genetic, or it might be a modeling effect.”
Gene research suggests that a region on chromosome 14 may be linked with compulsive hoarding in families with OCD. The study, carried out by a team from Johns Hopkins University School of Medicine in March 2007, analyzed samples from 999 OCD patients in 219 families. Families with two or more hoarding relatives showed a unique pattern on chromosome 14, whereas the other families’ OCD was linked to chromosome 3.
This was the third study to find genetic markers specifically associated with compulsive hoarding, according to Sanjaya Saxena, M.D., director of the University of California, San Diego, Obsessive-Compulsive Disorders Program.
In a letter to the editor of the American Journal of Psychiatry, she writes, “Other studies have confirmed that compulsive hoarding is strongly familial.” This research “adds to the mounting evidence indicating that compulsive hoarding is an etiologically discrete phenotype,” she believes.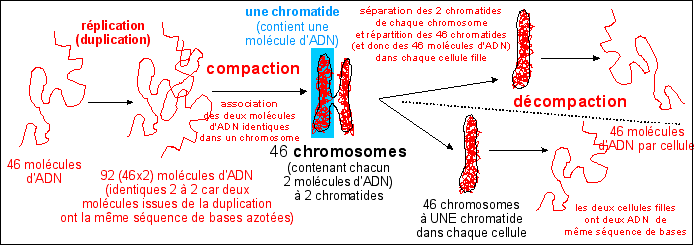
What’s more, brain imaging studies suggest that compulsive hoarding involves a specific type of brain activity. Patients have a different pattern of glucose metabolism in the brain than either healthy people or non-hoarding OCD patients.
Hoarding patients have significantly lower activity in the brain’s dorsal anterior cingulate cortex than non-hoarding OCD patients, and a different pattern of cognitive deficits was found, such as more difficulty making decisions and impaired decision-making.
Saxena concludes, “Compulsive hoarding syndrome appears to be a discrete entity, with a characteristic profile of core symptoms that are not strongly correlated with other OCD symptoms, distinct susceptibility genes, and unique neurobiological abnormalities that differ from those in non-hoarding OCD.”
OCD is a common feature of Tourette’s Syndrome, and this can include hoarding behavior, so a further gene study was undertaken by Heping Zhang, Ph.D. of Yale University School of Medicine and colleagues. Looking at the DNA of siblings with Tourette’s, the team found significant links to chromosome 4, 5, and 17.
Looking at the DNA of siblings with Tourette’s, the team found significant links to chromosome 4, 5, and 17.
“Something at chromosome 14 may be associated with hoarding,” says Randy Frost of Smith College. Writing in the Spring 2007 New England Hoarding Consortium Newsletter, he states, “This could be a dramatic breakthrough in our understanding of hoarding.
“However, it is important to note that these studies are all preliminary with relatively small samples that don’t fully represent the range of hoarding in the population. Furthermore, we also don’t yet understand just what traits might be heritable. Perhaps it is something that underlies hoarding, like decision-making problems, and not hoarding itself that is inherited.”
Much larger studies are needed, drawn from the entire population of people who hoard, not just those who are already diagnosed with OCD, he says. Frost is planning a project with experts from Johns Hopkins to answer the question more conclusively.
At present, his advice to people with hoarding tendencies in the family is to be open and honest with their children about the issue. “People who can recognize and talk about their own hoarding problems are much better able to control them than people who can’t.”
David F. Tolin, Ph.D., founder of the Anxiety Disorders Center at The Institute of Living in Hartford, CT, said that “for a condition like compulsive hoarding to come about you probably have to have a person who has a certain set of inherited characteristics. But biology is not destiny. Just because somebody has a genetic predisposition to develop a certain behavioral condition, that doesn’t mean they are doomed.”
References
Samuels, J. et al. Significant linkage to compulsive hoarding on chromosome 14 in families with obsessive-compulsive disorder: results from the OCD Collaborative Genetics Study. The American Journal of Psychiatry, Vol. 164, March 2007, pp. 493-99.
Saxena, S. Is Compulsive Hoarding a Genetically and Neurobiologically Discrete Syndrome? Implications for Diagnostic Classification. The American Journal of Psychiatry, Vol. 164, March 2007, pp. 380-84.
The American Journal of Psychiatry, Vol. 164, March 2007, pp. 380-84.
Saxena, S. et al. Cerebral Glucose Metabolism in Obsessive-Compulsive Hoarding. The American Journal of Psychiatry, Vol. 161, June 2004, pp. 1038-48.
Zhang, H. et al. Genomewide scan of hoarding in sib pairs in which both sibs have Gilles de la Tourette Syndrome. American Journal of Human Genetics, Vol. 70, April 2002, pp. 896-904.
Hoarding Newsletter (PDF)
Anxiety Disorders: Compulsive Hoarding
Significant linkage to compulsive hoarding on chromosome 14 in families with obsessive-compulsive disorder: results from the OCD Collaborative Genetics Study
Save citation to file
Format: Summary (text)PubMedPMIDAbstract (text)CSV
Add to Collections
- Create a new collection
- Add to an existing collection
Name your collection:
Name must be less than 100 characters
Choose a collection:
Unable to load your collection due to an error
Please try again
Add to My Bibliography
- My Bibliography
Unable to load your delegates due to an error
Please try again
Your saved search
Name of saved search:
Search terms:
Test search terms
Email: (change)
Which day? The first SundayThe first MondayThe first TuesdayThe first WednesdayThe first ThursdayThe first FridayThe first SaturdayThe first dayThe first weekday
Which day? SundayMondayTuesdayWednesdayThursdayFridaySaturday
Report format: SummarySummary (text)AbstractAbstract (text)PubMed
Send at most: 1 item5 items10 items20 items50 items100 items200 items
Send even when there aren't any new results
Optional text in email:
Create a file for external citation management software
Full text links
Atypon
Full text links
Case Reports
. 2007 Mar;164(3):493-9.
2007 Mar;164(3):493-9.
doi: 10.1176/ajp.2007.164.3.493.
Jack Samuels 1 , Yin Yao Shugart, Marco A Grados, Virginia L Willour, O Joseph Bienvenu, Benjamin D Greenberg, James A Knowles, James T McCracken, Scott L Rauch, Dennis L Murphy, Ying Wang, Anthony Pinto, Abby J Fyer, John Piacentini, David L Pauls, Bernadette Cullen, Steven A Rasmussen, Rudolf Hoehn-Saric, David Valle, Kung-Yee Liang, Mark A Riddle, Gerald Nestadt
Affiliations
Affiliation
- 1 Department of Psychiatry and Behavioral Sciences, Johns Hopkins University, School of Medicine, 600 N. Wolfe St., Meyer 4-181, Baltimore, MD 21287-7228, USA. [email protected]
- PMID: 17329475
- DOI: 10.
 1176/ajp.2007.164.3.493
1176/ajp.2007.164.3.493
Case Reports
Jack Samuels et al. Am J Psychiatry. 2007 Mar.
. 2007 Mar;164(3):493-9.
doi: 10.1176/ajp.2007.164.3.493.
Authors
Jack Samuels 1 , Yin Yao Shugart, Marco A Grados, Virginia L Willour, O Joseph Bienvenu, Benjamin D Greenberg, James A Knowles, James T McCracken, Scott L Rauch, Dennis L Murphy, Ying Wang, Anthony Pinto, Abby J Fyer, John Piacentini, David L Pauls, Bernadette Cullen, Steven A Rasmussen, Rudolf Hoehn-Saric, David Valle, Kung-Yee Liang, Mark A Riddle, Gerald Nestadt
Affiliation
- 1 Department of Psychiatry and Behavioral Sciences, Johns Hopkins University, School of Medicine, 600 N.
 Wolfe St., Meyer 4-181, Baltimore, MD 21287-7228, USA. [email protected]
Wolfe St., Meyer 4-181, Baltimore, MD 21287-7228, USA. [email protected]
- PMID: 17329475
- DOI: 10.1176/ajp.2007.164.3.493
Abstract
Objective: Individuals with obsessive-compulsive disorder (OCD) who have compulsive hoarding behavior are clinically different from other OCD-affected individuals. The objective of this study was to determine whether there are chromosomal regions specifically linked to compulsive hoarding behavior in families with obsessive-compulsive disorder.
Methods: The authors used multipoint allele-sharing methods to assess for linkage in 219 multiplex OCD families collected as part of the OCD Collaborative Genetics Study. The authors treated compulsive hoarding as the phenotype of interest and also stratified families into those with and without two or more relatives affected with compulsive hoarding.
The authors treated compulsive hoarding as the phenotype of interest and also stratified families into those with and without two or more relatives affected with compulsive hoarding.
Results: Using compulsive hoarding as the phenotype, there was suggestive linkage to chromosome 14 at marker D14S588 (Kong and Cox logarithm of the odds ratio [LOD] [KAC(all)=2.9]). In families with two or more hoarding relatives, there was significant linkage of OCD to chromosome 14 at marker C14S1937 (KAC(all)=3.7), whereas in families with fewer than two hoarding relatives, there was suggestive linkage to chromosome 3 at marker D3S2398 (KAC(all)=2.9).
Conclusions: The findings suggest that a region on chromosome 14 is linked with compulsive hoarding behavior in families with OCD.
Similar articles
-
Clinical correlates and genetic linkage of social and communication difficulties in families with obsessive-compulsive disorder: Results from the OCD Collaborative Genetics Study.
Samuels J, Shugart YY, Wang Y, Grados MA, Bienvenu OJ, Pinto A, Rauch SL, Greenberg BD, Knowles JA, Fyer AJ, Piacentini J, Pauls DL, Cullen B, Rasmussen SA, Stewart SE, Geller DA, Maher BS, Goes FS, Murphy DL, McCracken JT, Riddle MA, Nestadt G. Samuels J, et al. Am J Med Genet B Neuropsychiatr Genet. 2014 Jun;165B(4):326-36. doi: 10.1002/ajmg.b.32235. Epub 2014 May 4. Am J Med Genet B Neuropsychiatr Genet. 2014. PMID: 24798771
-
Genomewide linkage scan for obsessive-compulsive disorder: evidence for susceptibility loci on chromosomes 3q, 7p, 1q, 15q, and 6q.
Shugart YY, Samuels J, Willour VL, Grados MA, Greenberg BD, Knowles JA, McCracken JT, Rauch SL, Murphy DL, Wang Y, Pinto A, Fyer AJ, Piacentini J, Pauls DL, Cullen B, Page J, Rasmussen SA, Bienvenu OJ, Hoehn-Saric R, Valle D, Liang KY, Riddle MA, Nestadt G. Shugart YY, et al.
 Mol Psychiatry. 2006 Aug;11(8):763-70. doi: 10.1038/sj.mp.4001847. Epub 2006 Jun 6. Mol Psychiatry. 2006. PMID: 16755275
Mol Psychiatry. 2006 Aug;11(8):763-70. doi: 10.1038/sj.mp.4001847. Epub 2006 Jun 6. Mol Psychiatry. 2006. PMID: 16755275 -
Genomewide linkage analysis in Costa Rican families implicates chromosome 15q14 as a candidate region for OCD.
Ross J, Badner J, Garrido H, Sheppard B, Chavira DA, Grados M, Woo JM, Doo P, Umaña P, Fournier E, Murray SS, Mathews CA. Ross J, et al. Hum Genet. 2011 Dec;130(6):795-805. doi: 10.1007/s00439-011-1033-6. Epub 2011 Jun 21. Hum Genet. 2011. PMID: 21691774 Free PMC article.
-
Evidence for potential relationship between SLC1A1 and a putative genetic linkage region on chromosome 14q to obsessive-compulsive disorder with compulsive hoarding.
Liang KY, Wang Y, Shugart YY, Grados M, Fyer AJ, Rauch S, Murphy D, McCracken J, Rasmussen S, Cullen B, Hoehn-Saric R, Greenberg B, Pinto A, Knowles J, Piacentini J, Pauls D, Bienvenu O, Riddle M, Samuels J, Nestadt G.
 Liang KY, et al. Am J Med Genet B Neuropsychiatr Genet. 2008 Sep 5;147B(6):1000-2. doi: 10.1002/ajmg.b.30713. Am J Med Genet B Neuropsychiatr Genet. 2008. PMID: 18286588
Liang KY, et al. Am J Med Genet B Neuropsychiatr Genet. 2008 Sep 5;147B(6):1000-2. doi: 10.1002/ajmg.b.30713. Am J Med Genet B Neuropsychiatr Genet. 2008. PMID: 18286588 -
Genetics of obsessive-compulsive disorder: a research update.
Grados M, Wilcox HC. Grados M, et al. Expert Rev Neurother. 2007 Aug;7(8):967-80. doi: 10.1586/14737175.7.8.967. Expert Rev Neurother. 2007. PMID: 17678492 Review.
See all similar articles
Cited by
-
Bidirectional Behavioral Selection in Mice: A Novel Pre-clinical Approach to Examining Compulsivity.
Mitra S, Bult-Ito A. Mitra S, et al. Front Psychiatry. 2021 Sep 8;12:716619. doi: 10.3389/fpsyt.2021.716619. eCollection 2021.
 Front Psychiatry. 2021. PMID: 34566718 Free PMC article. Review.
Front Psychiatry. 2021. PMID: 34566718 Free PMC article. Review. -
Nordic OCD & Related Disorders Consortium: Rationale, design, and methods.
Mataix-Cols D, Hansen B, Mattheisen M, Karlsson EK, Addington AM, Boberg J, Djurfeldt DR, Halvorsen M, Lichtenstein P, Solem S, Lindblad-Toh K; Nordic OCD and Related Disorders Consortium (NORDiC), Haavik J, Kvale G, Rück C, Crowley JJ. Mataix-Cols D, et al. Am J Med Genet B Neuropsychiatr Genet. 2020 Jan;183(1):38-50. doi: 10.1002/ajmg.b.32756. Epub 2019 Aug 19. Am J Med Genet B Neuropsychiatr Genet. 2020. PMID: 31424634 Free PMC article.
-
Genetics of obsessive-compulsive disorder.
Purty A, Nestadt G, Samuels JF, Viswanath B. Purty A, et al. Indian J Psychiatry.
 2019 Jan;61(Suppl 1):S37-S42. doi: 10.4103/psychiatry.IndianJPsychiatry_518_18. Indian J Psychiatry. 2019. PMID: 30745675 Free PMC article. Review.
2019 Jan;61(Suppl 1):S37-S42. doi: 10.4103/psychiatry.IndianJPsychiatry_518_18. Indian J Psychiatry. 2019. PMID: 30745675 Free PMC article. Review. -
Are Mentalizing Abilities and Insight Related to the Severity of Obsessive-Compulsive Disorder.
İnanç L, Altıntaş M. İnanç L, et al. Psychiatry Investig. 2018 Sep;15(9):843-851. doi: 10.30773/pi.2018.05.02.2. Epub 2018 Aug 20. Psychiatry Investig. 2018. PMID: 30122030 Free PMC article.
-
Obsessive-Compulsive Disorder in a 19-Year-Old Female Adolescent With Turner Syndrome.
Moonga SS, Pinkhasov A, Singh D. Moonga SS, et al. J Clin Med Res. 2017 Dec;9(12):1026-1028. doi: 10.14740/jocmr3195w. Epub 2017 Nov 6. J Clin Med Res. 2017. PMID: 29163739 Free PMC article.
See all "Cited by" articles
Publication types
MeSH terms
Substances
Grant support
- K23-MH-64543/MH/NIMH NIH HHS/United States
- R01-MH-50214/MH/NIMH NIH HHS/United States
- RR00052/RR/NCRR NIH HHS/United States
Full text links
Atypon
Cite
Format: AMA APA MLA NLM
Send To
Genetic aspects of thyroid cancer | Garkavtseva
At present, there is no doubt that malignant neoplasms are based on damage to the genetic apparatus in germinal (sex) or somatic cells, making these cells sensitive to the effects of environmental carcinogenic factors that can activate the process of malignancy. Cancer can be hereditary or non-hereditary depending on whether the initial mutation occurred in a sexual or somatic cell. The genes responsible for transformation are normal cellular genes involved in the control of cell division, growth, and differentiation, but the change in these genes ultimately leads to uncontrolled cell division. In hereditary forms of the tumor, the initial changes (mutations) that occur in germ cells will be detected in all cells, including peripheral blood cells. However, the initial mutation in the sex or somatic cell is not sufficient for the development of a tumor. In the event of a similar mutation of an alternative gene in the homologous chromosome, a malignant transformation may develop.
Cancer can be hereditary or non-hereditary depending on whether the initial mutation occurred in a sexual or somatic cell. The genes responsible for transformation are normal cellular genes involved in the control of cell division, growth, and differentiation, but the change in these genes ultimately leads to uncontrolled cell division. In hereditary forms of the tumor, the initial changes (mutations) that occur in germ cells will be detected in all cells, including peripheral blood cells. However, the initial mutation in the sex or somatic cell is not sufficient for the development of a tumor. In the event of a similar mutation of an alternative gene in the homologous chromosome, a malignant transformation may develop.
This also applies to hereditary forms of thyroid cancer.
According to modern concepts, thyroid cancer is a heterogeneous disease in terms of histogenesis, tumor structure, and etiological factors.
A-cell neoplasms of the thyroid gland (papillary and follicular thyroid cancer)
or papillary thyroid cancer (PTC) among first-degree relatives [16]. Familial variants of PTC and follicular thyroid cancer (FTC) account for 3-7% of all cases of these tumors. In such families, 2–4 relatives affected by similar tumors are detected, and in some families up to 8 affected relatives [5]. The clinical picture and prognosis for familial PTC do not differ from those for sporadic forms of this disease. However, population studies have shown that in families where several cases of these forms of cancer have already been detected, the risk of its occurrence in relatives is 8.6 times greater than in the control group [9]. This risk is increased if both parents have thyroid cancer. In addition, the involvement of hereditary factors in the development of these forms of cancer was confirmed by observations of papillary adenocarcinoma in monozygotic twins, followed by the development of bilateral breast cancer in them. The molecular defect responsible for the development of these forms of diseases is still unknown, but an active search for candidate genes is underway.
Familial variants of PTC and follicular thyroid cancer (FTC) account for 3-7% of all cases of these tumors. In such families, 2–4 relatives affected by similar tumors are detected, and in some families up to 8 affected relatives [5]. The clinical picture and prognosis for familial PTC do not differ from those for sporadic forms of this disease. However, population studies have shown that in families where several cases of these forms of cancer have already been detected, the risk of its occurrence in relatives is 8.6 times greater than in the control group [9]. This risk is increased if both parents have thyroid cancer. In addition, the involvement of hereditary factors in the development of these forms of cancer was confirmed by observations of papillary adenocarcinoma in monozygotic twins, followed by the development of bilateral breast cancer in them. The molecular defect responsible for the development of these forms of diseases is still unknown, but an active search for candidate genes is underway. Recently published data on the localization of the gene on the short arm of chromosome 19(19p13.2) [6], responsible for the development of a thyroid tumor, and a gene on the long arm of chromosome 14 (14q31), responsible for the development of multinodular goiter [12]. In addition, a gene is localized on the long arm of chromosome 1 (lq21) associated with the familial form of PTC in combination with kidney cancer [14].
Recently published data on the localization of the gene on the short arm of chromosome 19(19p13.2) [6], responsible for the development of a thyroid tumor, and a gene on the long arm of chromosome 14 (14q31), responsible for the development of multinodular goiter [12]. In addition, a gene is localized on the long arm of chromosome 1 (lq21) associated with the familial form of PTC in combination with kidney cancer [14].
Hereditary variants also include the so-called "cancer families" syndromes, in which, along with tumors of the mammary glands, intestines, stomach, endometrium and other neoplasias, PTC or FTC are detected in several members of the same family. The most well-known is the association of A-cell thyroid cancer with familial colon polyposis and its variants such as Gardner's syndrome, Turkot syndrome. Thyroid cancer in Gardner's syndrome (colon adenomatosis, skin fibromas and epidermoid cysts) occurs with a frequency of 1-3% of cases. Women of young age — 20 — 30 years (the ratio of women and men makes 10:1) are more often surprised. In most cases, the tumor associated with these syndromes is multicentric and precedes the detection of intestinal polyps in 2/3 of cases. These features indicate the advisability of periodic examination of the colon in young patients with thyroid cancer in order to detect intestinal adenomatosis and, conversely, in patients with familial colon polyposis, a thorough examination of the thyroid gland is recommended. A number of patients with PTC associated with these syndromes have mutations in the APC (adenomatous poliposis coli) gene located in the long arm of chromosome 5 (5q21) and a number of other genes [17].
In most cases, the tumor associated with these syndromes is multicentric and precedes the detection of intestinal polyps in 2/3 of cases. These features indicate the advisability of periodic examination of the colon in young patients with thyroid cancer in order to detect intestinal adenomatosis and, conversely, in patients with familial colon polyposis, a thorough examination of the thyroid gland is recommended. A number of patients with PTC associated with these syndromes have mutations in the APC (adenomatous poliposis coli) gene located in the long arm of chromosome 5 (5q21) and a number of other genes [17].
Predisposition to the occurrence of Cowden's syndrome, in which there is an increased risk of developing PTC, is due to a hereditary mutation in the PTEN gene, which, like the APC gene, belongs to oncosuppressor genes.
As for sporadic PTC, today it is already known that this disease is based on somatic mutations affecting the RET gene, followed by the formation of chimeric oncogenes encoding cytosolic chimeric proteins [4].![]() The ability of the thyroid tissue to accumulate iodine and its related elements underlies the increase in the frequency of chromosomal rearrangements in thyroid cells and the activation of the RET proto-oncogene under the influence of their radioactive isotopes. This explains the increase in the incidence of PTC after the Chernobyl disaster.
The ability of the thyroid tissue to accumulate iodine and its related elements underlies the increase in the frequency of chromosomal rearrangements in thyroid cells and the activation of the RET proto-oncogene under the influence of their radioactive isotopes. This explains the increase in the incidence of PTC after the Chernobyl disaster.
C-cell neoplasms of the thyroid gland (medullary thyroid cancer)
The largest proportion of hereditary cases (20–30%) is observed in medullary thyroid cancer (MTC) [1, 2], which is the result of inheritance of mutations in the RET proto-oncogene.
For the first time, the histological features of MTC, in particular, the presence of amyloid in the stroma, were described by J. Hazard et al. [ten]. They emphasized the need to isolate this form of the tumor due to the peculiarities of its histological structure and clinical course, which occupies an intermediate position in prognosis between highly differentiated and anaplastic thyroid cancer.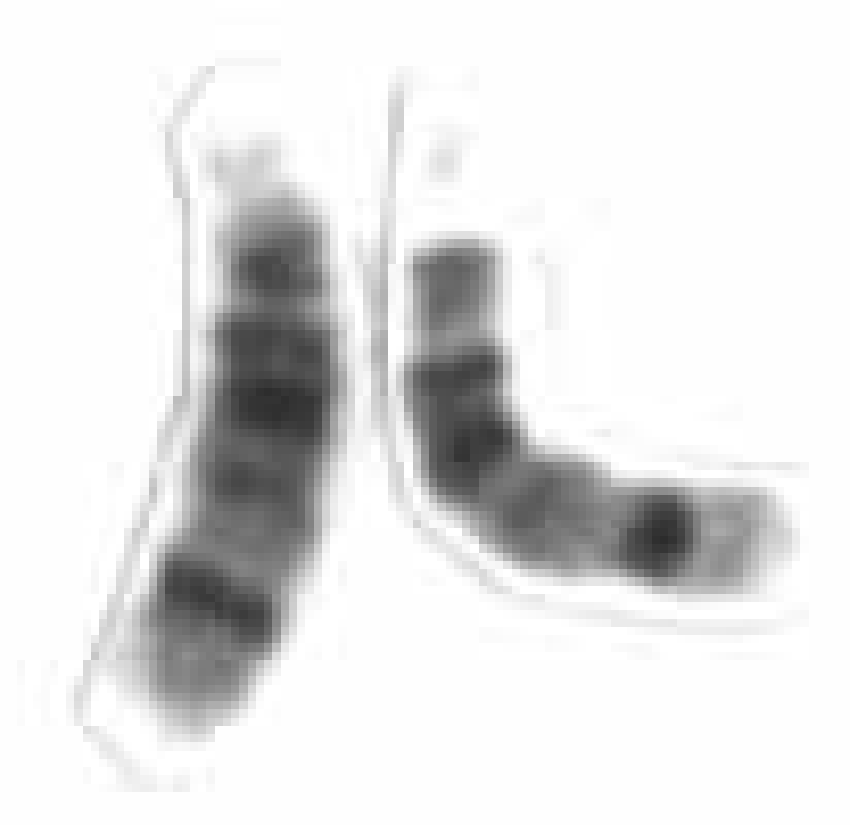 The same authors introduced the term "medullary thyroid cancer". At 1963 R. Manning et al. [15] described the combination of pheochromocytoma, MTC and hyperparathyroidism. The genetic nature of this syndrome was soon established. Subsequently, cases of another hereditary syndrome were described, including MTC, pheochromocytoma, mucosal neuroma, neurofibromatosis, and a morphan-like phenotype [3]. In 1968 A. Steiner et al. [22] proposed the term “multiple endocrine neoplasia syndrome” type 2, MEN-2, to designate these tumor combinations (MEN-1, which includes tumor lesions of the parathyroid glands, pituitary gland, and the hormone-producing part of the pancreas, was described earlier). Subsequently, MEN-2 was subdivided into MEN-2A (Cipple's syndrome) and MEN-2B (Gorlin's syndrome).
The same authors introduced the term "medullary thyroid cancer". At 1963 R. Manning et al. [15] described the combination of pheochromocytoma, MTC and hyperparathyroidism. The genetic nature of this syndrome was soon established. Subsequently, cases of another hereditary syndrome were described, including MTC, pheochromocytoma, mucosal neuroma, neurofibromatosis, and a morphan-like phenotype [3]. In 1968 A. Steiner et al. [22] proposed the term “multiple endocrine neoplasia syndrome” type 2, MEN-2, to designate these tumor combinations (MEN-1, which includes tumor lesions of the parathyroid glands, pituitary gland, and the hormone-producing part of the pancreas, was described earlier). Subsequently, MEN-2 was subdivided into MEN-2A (Cipple's syndrome) and MEN-2B (Gorlin's syndrome).
MEN-2 is inherited in an autosomal dominant manner. The clinical features of this syndrome are pronounced phenotypic polymorphism, which is manifested by three types of the disease: MEN-2A, MEN-2B and familial MTC.
Patients with MEN-2A are characterized by the presence of MTC, as well as pheochromocytoma (50% of patients) and parathyroid hyperplasia (20–30% of patients) [20]. MEN-2B has the same symptoms as MEN-2A but does not have parathyroid hyperplasia. Patients with MEN-2B are also characterized by a morphan-like physique, mucosal neurinomas, and neurofibromatosis [20]. In addition, familial MTC can occur in isolation. MTC is formed from calcitonin-producing parafollicular cells (or C-cells) of the thyroid gland, which make up less than 1% of the total cell mass of this organ, are derivatives of the neural crest and have features characteristic of the APUD system. C cells secrete the hormone calcitonin, which consists of 32 amino acid residues [7] and has several immunoreactive forms. MTC accounts for 4–20% of all carcinomas of this localization. The age of patients varies from 13 to 83 years, the average age is 44 ± 16 years for patients with the sporadic form, 32 ± 1 years for those affected by the MEN-2A syndrome, and 25 ± 7 years for the MEN-2B syndrome [20].
The genetic basis of diseases with MEN-2 syndromes in almost all cases studied are inherited missense mutations in the RET protooncogene [11]. The RET (RE-arranged during Transfection) gene is located on the long arm of chromosome 10 (10qll.2). It includes 21 exons. The products of the RET gene are tyrosine kinases (TKs) of the receptor type (polypeptides consisting of 1072–1114 amino acids), which are involved in the control of proliferation, migration, and/or differentiation of neural crest cells [23].
In humans, the RET gene is expressed in normal and neoplastic tissues of neuroendocrine origin, including parafollicular C cells and MTC, adrenal medulla and pheochromocytoma, neuroblastomas, peripheral nerves and their tumors.
Most RET mutations found in patients with MEN-2 syndromes show clustering. They are localized in one of two regions of the PET proto-oncogene. A single point mutation at codon 918 (exon 16) was found in more than 95% of families with MEN-2B [R]. This mutation (ATG->ACG) results in the replacement of methionine by threonine in the cytoplasmic tyrosine kinase domain of the RET protein. Missense mutations in 1 of 5 cysteine codons - 609, 611, 618, 620 (exon 10) or 634 (exon 11) - in the extracellular domain of TK were identified in more than 95% of families with MEN-2A and in 70-80% with familial MTC [13].
This mutation (ATG->ACG) results in the replacement of methionine by threonine in the cytoplasmic tyrosine kinase domain of the RET protein. Missense mutations in 1 of 5 cysteine codons - 609, 611, 618, 620 (exon 10) or 634 (exon 11) - in the extracellular domain of TK were identified in more than 95% of families with MEN-2A and in 70-80% with familial MTC [13].
In patients with sporadic forms of MTC, somatic mutations in the RET gene have also been identified, which overlap with mutations in MEN-2.
It has now been established that a number of mutations in the RET proto-oncogene affecting 1 of 5 cysteines in the cysteine-rich region of the extracellular domain of TK lead to constitutive activation of TK as a result of the formation of covalently bound homodimers of the RET protein [19|.
MEN-2B mutation in codon 918 (tyrosine kinase domain) also leads to constitutive activation of TK, but without dimerization of the latter. In addition, this mutation changes the substrate specificity of the enzyme [19], which leads to autophosphorylation of tyrosine residues of TK at new sites, as well as to the phosphorylation of some new cellular proteins.
Mutations in RET oncogenes can lead to fibroblast transformations [19].
Thus, mutations associated with MEN-2 syndromes transform the normal RET proto-oncogene into a dominant transforming oncogene whose products have constitutive tyrosine kinase activity and increased phosphorylation capacity. In the case of the mutation detected by MEN-2B, MCs exhibit altered substrate specificity. The mechanism of malignant RET transformation of C cells in vivo is currently unknown.
The material accumulated to date on MEN-2 makes it possible to establish a relationship between certain mutations found in patients and their phenotype. Thus, when examining the largest sample of families with MEN-2, collected by the International Consortium on RET Mutations (477 families), a close relationship was established between the presence of one of the mutations in codon 634 and the presence of pheochromocytoma and parathyroid hyperplasia [8]. In the surveyed Russian family with MEN-2A (Cys 634 Arg mutation), we also found the following relationship: the presence of pheochromocytoma was established for members of the 1/2, 2/2, and 2/3 pedigree (Fig. 1).
The results of the analysis of studies on the genetics of thyroid cancer can be summarized as follows. The results of research in this area indicate the undoubted presence of genetically determined forms of thyroid cancer, the proportion of which in the structure of the oncopathology of this organ depends primarily on the histogenesis of the tumor. Thus, works devoted to A-cell carcinoma are mostly descriptive, presenting individual observations of family cases of this pathology, as well as attempts to search for biochemical and molecular markers. At the same time, genetic heterogeneity and clinical polymorphism are noted in C-cell thyroid cancer. Based on these data, the principles of medical genetic counseling were developed, aimed at clarifying the prognosis in the offspring of affected individuals, early diagnosis and prevention of the disease.
As a method for identifying hereditary forms of thyroid cancer and forming risk groups that are subject to in-depth clinical examination in order to identify C-cell thyroid pathology in them, we have proposed a program (Fig. 2) based on the genetic heterogeneity of thyroid cancer, the occurrence which, based on the Knudson two-mutation theory, is determined by 1 of 3 possible options: 1) inheritance of a mutation in germ cells, resulting in a genetically determined predisposition to medullary cancer, manifested in the form of C-cell hyperplasia with hypercalcitoninemia (sometimes hidden), which is realized in cancer as a result of a second (somatic) mutation in thyroid tissue cells; 2) "de novo" mutation in the germinal cells of one of the parents. In this case, the phenotypic manifestations are similar to the previous option in the absence of the disease or predisposition to it in the parents; 3) two somatic mutations "de novo", i.e. mutations in the C-cells of the thyroid gland, which are not inherited.
The most important from the point of view of the formation of genetic risk groups is the first variant, which can be detected in a patient by determining the level of calcitonin in the blood serum and detecting a mutation in the RET gene. The sensitivity of calcitonin determination (especially after stimulation with pentagastrin) is so great that some researchers recommend performing thyroid surgery even without obvious clinical signs of the disease, only on the basis of an increase in basal or stimulated hormone levels in the blood serum with a positive family history. In the analysis of 14 works devoted to this still controversial issue, the following data were obtained: thyroidectomy was performed in 80 individuals from MTC families. Indications for surgery were determined by the presence of hypercalcitoninemia (in some cases after stimulation). Post- and preoperative histological examination made it possible in 61 cases to detect microfoci of MTC that were not detected by clinical or other examination methods; 18 patients were diagnosed with C-cell hyperplasia, and only in 1 case no pathological changes were found [1, 2].
Currently, in the presence of an elevated level of calcitonin and the detection of a mutation in the RET gene, it is recommended to conduct a targeted examination of all relatives of the 1st degree of kinship to identify persons who are carriers of the pathological gene (the analysis is carried out with the consent of the subject). In such a situation, cancer must be considered as a Mendelian trait, inherited in an autosomal dominant manner, and the risk to the offspring of patients is approximately 50%, given the high penetrance. For siblings of patients and siblings of parents, the same probability of getting sick remains, so they must be included in the high-risk group with constant dispensary observation and mandatory determination of the level of calcitonin after stimulation with pentagastrin and detection of RET gene mutations.
If a mutation in the RET gene is detected, then an annual test with pentagastrin is recommended from the age of 5-6 years. With a high content of calcitonin, total thyroidectomy is recommended at the age of 10-13 years. Approximately 50% of children at this age have microscopic foci of MTC. Using this approach allows us to talk about a 90% cure for children [21]. Such an incomplete cure by many authors is explained, on the one hand, by the presence of undiagnosed metastases in the cervical lymph nodes, and on the other hand, by the incomplete removal of the thyroid gland and part of the C-cells, which are the source of MTC recurrence.
Another approach to the treatment and follow-up of patients with hereditary MTC and MEN-2A and MEN-2B syndromes is to perform thyroidectomy based only on the detection of an inherited RET gene mutation, since more than 90% of children with this mutation will develop MTC sooner or later . In this case, radical thyroidectomy before the age of 6 years is recommended, followed by replacement therapy with levothyroxine. Such prophylactic surgical treatment is quite justified, since the likelihood of C-cell transformation and metastasis at this age is excluded. Filed by V. Pasini et al. [18], genetic screening is usually positively perceived by family members, and early and radical surgical treatment of a child is usually supported by parents.
The pedigree of the family observed by us in the Russian Cancer Research Center can serve as an illustration of what has been said (see Fig. 1). In patient (3/2), the clinically established diagnosis of MEN-2A was confirmed by molecular studies: during the analysis of exon 11 of the RET gene, we found the known and most frequent TGC-»CGC mutation in codon 634 in the heterozygous state in MEN-2A. The identified mutation leads to the replacement of the cysteine amino acid residue in the cysteine-rich extracellular tyrosine kinase domain of RET with arginine. A similar mutation was identified in the patient's mother (2/3) and cousin (3/1). The patient (like his 91-year-old cousin) underwent prophylactic thyroidectomy. Histological examination of the surgical material of the proband revealed 4 foci of MTC.
Thus, the identification of RET mutations in family members with MEN-2 allows early (preclinical) diagnosis of the disease, i.e., to identify potential patients before they develop clinical or biochemical signs of medullary cancer and thereby reduce the risk of potentially lethal disease. It should be emphasized that the early molecular diagnosis of MEN-2 is highly sensitive and accurate: so far there have been no reports of the development of MTC in non-RET-mutant individuals from MEN-2 families with known RET mutations. In this regard, the introduction of DNA diagnostics into the practice of Russian oncologists seems to be absolutely necessary.
1. Garkavtseva R. F., Sotnikova E. N., Kazubskaya T. P. et al // Medicine and health care: Review information. - M., 1990. - No. 2.
2. Lisnyansky I. E. // VNIIMI. Express information. Ser.: Med. genetics. - 1986. - No. 4. - S. 1-29.
3. Shimke RN Genetics and cancer in humans. M., 1981.
4. Bongarzone /., Butti M. G., Coronelli S. et al. // Cancer Res. - 1994. - Vol. 54. - P. 2979-2985.
5. Burgess J. R., Duffield A., Wilkinson S J. et al // J. Clin. Endocrinol. Metab. - 1997. - Vol. 82. - P. 345-348.
6. Canzian E, Amati P., Harach H. R. et al. // Ant. J. Hum. Genet. - 1998. - Vol. 63. - P. 1743-1748.
7. Defies L. J. I/ S. Karger. Basel, 1983.
8. Eng C., Clayton D., Schuffenecker I. et al. // J. A. M. A. - 1994. - Vol. 276. - P. 1575-1579.
9. Goldgar D. E., Easton D. E, Cannon Albright L. A., Skolnick
10. M. H. // J. Natl. Cancer Inst. - 1994. - Vol. 86. - P. 1600-1608.
11. Hazard J. D., Hawk W. A., Crile G. J. // J. Clin. Endocrinol. - 1959. - Vol. 19, No. 4. - P. 152-161.
12. Hofstra R. M. W., Landsvater R M., Ceccherini I. et al. // Nature. - 1994. - Vol. 367. - P. 375-376.
13. Lesueur E, Stark M., Tocco T. et al. // J. Clin. Endocrinol. Metab. - 1999. - Vol. 84. - P. 2157-2162.
14. Lips C. J. M., Landsvater R. M., Hoppener J. W. M. et al. // N. Engl. J. Med. -1994. - Vol. 331. - P. 828-835.
15. Malchoff C. D., Satfarazi M., Tendler B. et al. // J. Clin. Endocrinol. Metab. -2000. - Vol. 85. - P. 1758-1764.
16. Manning P. C., Nolnar G. D., Diack B M. et al. // N. Engl. J. Med. - 1963. - Vol. 268, No. 12. - P. 68-72.
17. Nemec J., Soumar J., Zamrazil V. et al. // Oncology. - 1975. - Vol. 32, No. 3-4. - P. 151-157.
18. Nishisho L, Nakamura Y., Mivoshi Y. et al. // Science. - 1991. - Vol. 53.-P. 665-669.
19. Pasini B., Hofstra R. M. W., Yin L. et al. // Oncogene. - 1994. - Vol. 11. - P. 1737-1743.
20. Saad M. V., Ordonez N. G., Rashid R. K. et al. // medicine. - 1984.-Vol. 63, No. 6. - P. 319-342.
21. Santoro M., Carlomagno E. Romano A. et al. // Science. - 1995. - Vol. 267. - P. 381-383.
22. Schimke R. // Ann. Rev. Med. - 1984. - Vol. 35. - P. 25-31.
23. Steiner A. L., Goodman A. D., Powers S. R. // Medicine. - 1968. - Vol. 47, No. 5. - P. 371-409.
24. Takahashi M., Burna Y, Iwamoto T. et al. // Oncogene. - 1988. - Vol. 3. - P. 571-578.
INVITRO. Hereditary diseases, find out the prices for tests and take them in Moscow
- Systemic connective tissue diseases
- Rheumatoid arthritis, joint damage
- Antiphospholipid syndrome
- Vasculitis and kidney damage
- Autoimmune lesions of the gastrointestinal tract. Celiac disease
- Autoimmune liver diseases
- Neurological autoimmune diseases
- Autoimmune endocrinopathies
- Autoimmune skin diseases
- Lung and heart diseases
- Hereditary metabolic diseases
- Screening of newborns to detect hereditary metabolic diseases
- Additional tests (after screening and specialist consultation)
- Water and soil quality survey
- Water quality survey
- New tests
- Obtaining results
- Additional research orders
- Medical consultant service
- Professional position
- Venous blood for analysis
- Tumor markers.