Rett syndrome definition
Rett syndrome - Symptoms and causes
Overview
Rett syndrome is a rare genetic neurological and developmental disorder that affects the way the brain develops. This disorder causes a progressive loss of motor skills and language. Rett syndrome primarily affects females.
Most babies with Rett syndrome seem to develop as expected for the first six months of life. These babies then lose skills they previously had — such as the ability to crawl, walk, communicate or use their hands.
Over time, children with Rett syndrome have increasing problems with the use of muscles that control movement, coordination and communication. Rett syndrome can also cause seizures and intellectual disabilities. Unusual hand movements, such as repetitive rubbing or clapping, replace purposeful hand use.
Although there's no cure for Rett syndrome, potential treatments are being studied. Current treatment focuses on improving movement and communication, treating seizures, and providing care and support for children and adults with Rett syndrome and their families.
Products & Services
- Book: Mayo Clinic Family Health Book, 5th Edition
- Newsletter: Mayo Clinic Health Letter — Digital Edition
Symptoms
Babies with Rett syndrome usually are born after an uncomplicated pregnancy and delivery. Most infants with Rett syndrome seem to grow and behave as expected for the first six months. After that, signs and symptoms start to appear.
The most pronounced changes generally occur at 12 to 18 months of age, over a period of weeks or months. Symptoms and their severity vary greatly from child to child.
The main signs and symptoms include:
- Slowed growth. Brain growth slows after birth. Smaller than usual head size (microcephaly) is sometimes the first sign that a child has Rett syndrome. As children get older, there is delayed growth in other parts of the body.
- Loss of movement and coordination abilities. The first signs often include reduced hand control and a decreasing ability to crawl or walk.
 At first, this loss of abilities occurs rapidly, and then it continues more gradually. Eventually muscles become weak or stiff, with unusual movement and positioning.
At first, this loss of abilities occurs rapidly, and then it continues more gradually. Eventually muscles become weak or stiff, with unusual movement and positioning. - Loss of communication abilities. Children with Rett syndrome typically begin to lose the ability to speak, to make eye contact and to communicate in other ways. They may become disinterested in other people, toys and their surroundings. Some children have rapid changes, such as a sudden loss of language. Over time, children may gradually regain eye contact and develop nonverbal communication skills.
- Unusual hand movements. Children with Rett syndrome usually develop repetitive, purposeless hand movements, which differ from child to child. Hand movements may include hand-wringing, squeezing, clapping, tapping or rubbing.
Other signs and symptoms can include:
- Unusual eye movements. Children with Rett syndrome tend to have unusual eye movements, such as intense staring, blinking, crossed eyes or closing one eye at a time.

- Breathing problems. These include breath holding, rapid breathing (hyperventilation), forcefully blowing out air or saliva, and swallowing air. These problems tend to occur during waking hours. Other breathing disturbances such as shallow breathing or short periods of stopping breathing (apnea) can occur during sleep.
- Irritability and crying. Children with Rett syndrome may become increasingly agitated and irritable as they get older. Periods of crying or screaming may begin suddenly, for no apparent reason, and last for hours. Some children may experience fears and anxiety.
- Other unusual behaviors. These may include, for example, sudden, odd facial expressions and long bouts of laughter, hand licking, and grasping of hair or clothing.
- Intellectual disabilities. Loss of skills may be connected to losing the ability to think, understand and learn.
- Seizures.
 Most people who have Rett syndrome experience seizures at some time during their lives. Multiple seizure types may occur and are associated with changes on an electroencephalogram (EEG).
Most people who have Rett syndrome experience seizures at some time during their lives. Multiple seizure types may occur and are associated with changes on an electroencephalogram (EEG). - Sideways curvature of the spine (scoliosis). Scoliosis is common with Rett syndrome. It typically begins between 8 and 11 years of age and progresses with age. Surgery may be required if the curvature is severe.
- Irregular heartbeat. This is a life-threatening problem for many children and adults with Rett syndrome and can result in sudden death.
- Sleep disturbances. Problems with sleep patterns can include irregular sleep times, falling asleep during the day and being awake at night, or waking in the night with crying or screaming.
- Other symptoms. A variety of other symptoms can occur, such as a decreased response to pain; small hands and feet that are usually cold; problems with chewing and swallowing; problems with bowel function; and teeth grinding.

Stages of Rett syndrome
Rett syndrome is commonly divided into four stages:
- Stage 1: Early onset. Signs and symptoms are subtle and easily overlooked during the first stage, which starts between 6 and 18 months of age. Stage 1 can last for a few months or a year. Babies in this stage may show less eye contact and start to lose interest in toys. They may also have delays in sitting or crawling.
- Stage 2: Rapid deterioration. Starting between 1 and 4 years of age, children lose the ability to perform skills they previously had. This loss can be rapid or more gradual, occurring over weeks or months. Symptoms of Rett syndrome occur, such as slowed head growth, abnormal hand movements, hyperventilating, screaming or crying for no apparent reason, problems with movement and coordination, and a loss of social interaction and communication.
- Stage 3: Plateau. The third stage usually begins between the ages of 2 and 10 years, and it can last for many years.
 Although problems with movement continue, behavior may slightly improve, with less crying and irritability, and there may be some improvement in hand use and communication. Seizures may begin in this stage and generally don't occur before the age of 2.
Although problems with movement continue, behavior may slightly improve, with less crying and irritability, and there may be some improvement in hand use and communication. Seizures may begin in this stage and generally don't occur before the age of 2. - Stage 4: Late motor deterioration. This stage usually begins after the age of 10 and can last for years or decades. It's marked by reduced mobility, muscle weakness, joint contractures and scoliosis. Understanding, communication and hand skills generally remain stable or improve slightly, and seizures may occur less often.
When to see a doctor
Signs and symptoms of Rett syndrome can be subtle in the early stages. See your child's health care provider right away if you begin to notice physical problems or changes in behavior after what appears to be typical development. Problems or changes may include:
- Slowed growth of your child's head or other parts of the body
- Decreased coordination or mobility
- Repetitive hand movements
- Decreasing eye contact or loss of interest in usual play
- Delayed language development or loss of previous language abilities
- Any clear loss of previously gained milestones or skills
Request an appointment
Causes
Rett syndrome is a rare genetic disorder. Classic Rett syndrome, as well as several variants (atypical Rett syndrome) with milder or more-severe symptoms, occur based on several specific genetic changes (mutations).
Classic Rett syndrome, as well as several variants (atypical Rett syndrome) with milder or more-severe symptoms, occur based on several specific genetic changes (mutations).
The genetic changes that cause Rett syndrome occur randomly, usually in the MECP2 gene. Very few cases of this genetic disorder are inherited. The genetic changes appear to result in problems with the protein production critical for brain development. However, the exact cause is not fully understood and is still being studied.
Rett syndrome in males
Because males have a different chromosome combination from females, males who have the genetic changes that cause Rett syndrome are affected in devastating ways. Most of them die before birth or in early infancy.
A very small number of males have a different genetic change that results in a less destructive form of Rett syndrome. Similar to females with Rett syndrome, these males are likely to live to adulthood, but they're still at risk of a number of intellectual and developmental problems.
Risk factors
Rett syndrome is rare. The genetic changes known to cause the disease are random, and no risk factors have been identified. In a very small number of cases, inherited factors — for instance, having close family members with Rett syndrome — may play a role.
Complications
Complications of Rett syndrome include:
- Sleep problems that cause significant sleep disruption to the person with Rett syndrome and family members.
- Difficulty eating, leading to poor nutrition and delayed growth.
- Bowel and bladder problems, such as constipation, gastroesophageal reflux disease (GERD), bowel or urinary incontinence, and gallbladder disease.
- Pain that may accompany problems such as gastrointestinal issues or bone fractures.
- Muscle, bone and joint problems.
- Anxiety and problem behavior that may hinder social functioning.
- Needing lifelong care and assistance with activities of daily living.

- Shortened life span. Although most people with Rett syndrome live into adulthood, they may not live as long as the average person because of heart problems and other health complications.
Prevention
There's no known way to prevent Rett syndrome. In most cases, the genetic changes that cause the disorder occur spontaneously. Even so, if you have a child or other family member with Rett syndrome, you may want to ask your health care provider about genetic testing and genetic counseling.
By Mayo Clinic Staff
Related
Associated Procedures
Products & Services
Rett Syndrome | National Institute of Neurological Disorders and Stroke
What is Rett syndrome?
Rett syndrome is a neurodevelopmental disorder characterized by typical early growth and development followed by a slowing of development, loss of functional use of the hands, distinctive hand movements, slowed brain and head growth, problems with walking, seizures, and intellectual disability.
The course of Rett syndrome, including the age of onset and the severity of symptoms, varies from child to child. Before the symptoms begin, however, the child generally appears to grow and develop, although there are often subtle abnormalities even in early infancy, such as:
- Loss of muscle tone (hypotonia)
- Difficulty feeding
- Jerkiness in limb movements
Gradually, mental and physical symptoms appear. Other early symptoms may include:
- Problems crawling or walking
- Diminished eye contact
The loss of functional use of the hands is followed by compulsive hand movements such as wringing and washing. The onset of this period of regression is sometimes sudden.
Apraxia, which is the inability to perform motor functions, is perhaps the most severely disabling feature of Rett syndrome, interfering with every body movement, including eye gaze and speech.
Children with Rett syndrome often exhibit autistic-like behaviors in the early stages. Other symptoms may include:
Other symptoms may include:
- Walking on the toes
- Sleep problems
- A wide-based gait
- Teeth grinding and difficulty chewing
- Slowed growth
- Seizures
- Cognitive disabilities
- Breathing difficulties while awake such as hyperventilation, apnea (breath holding), and air swallowing
Who is more likely to get Rett syndrome?
Rett syndrome is estimated to affect all racial and ethnic groups worldwide. It affects girls almost exclusively. Prenatal testing is available for families with an affected daughter who has an identified MECP2 mutation.
Genetic testing is also available for sisters of girls with Rett syndrome who have an identified MECP2 mutation to determine if they are carriers of the disorder.
The MECP2 gene is found on the X chromosome, one of the two sex chromosomes. Girls have two X chromosomes, but only one is active in any given cell. This means that in a girl with Rett syndrome only a portion of the cells in the nervous system will use the defective
Nearly all cases of Rett syndrome are caused by a mutation in the methyl CpG binding protein 2, or MECP2 (pronounced meck-pea-two) gene.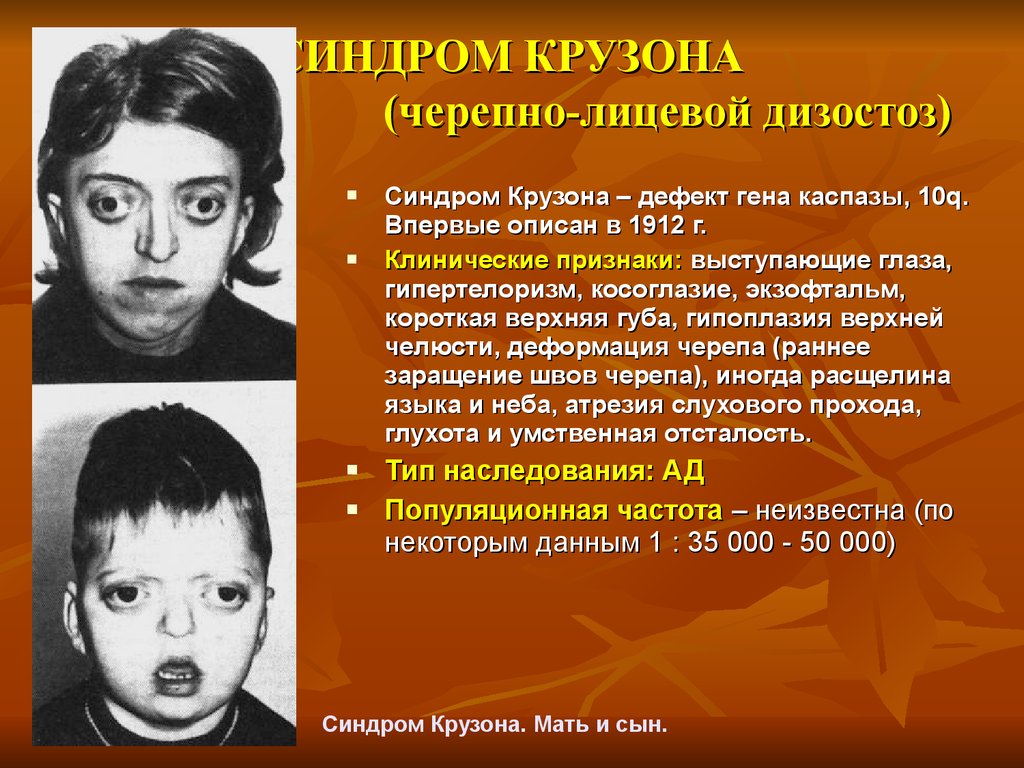 The MECP2 gene contains instructions for the synthesis of a protein called methyl cytosine binding protein 2 (MeCP2), which is needed for brain development and acts as one of the many biochemical switches that can either increase gene expression or tell other genes when to turn off and stop producing their own unique proteins. Because theMECP2 gene does not function properly in individuals with Rett syndrome, insufficient amounts or structurally abnormal forms of the protein are produced and can cause other genes to be abnormally expressed.
The MECP2 gene contains instructions for the synthesis of a protein called methyl cytosine binding protein 2 (MeCP2), which is needed for brain development and acts as one of the many biochemical switches that can either increase gene expression or tell other genes when to turn off and stop producing their own unique proteins. Because theMECP2 gene does not function properly in individuals with Rett syndrome, insufficient amounts or structurally abnormal forms of the protein are produced and can cause other genes to be abnormally expressed.
Not everyone who has an MECP2 mutation has Rett syndrome. Scientists have identified mutations in the CDKL5 and FOXG1 genes in individuals who have atypical or congenital Rett syndrome, but they are still learning how those mutations cause the disorder. Scientists believe the remaining cases may be caused by partial gene deletions, mutations in other parts of the MECP2 gene, or additional genes that have not yet been identified, and they continue to look for other causes.
Although Rett syndrome is a genetic disorder, less than one percent of recorded cases are inherited or passed from one generation to the next. Most cases are spontaneous, which means the mutation occurs randomly. However, in some families of individuals affected by Rett syndrome, there are other female family members who have a mutation of their MECP2 gene.
How is Rett syndrome diagnosed and treated?
Diagnosing Rett syndrome
Doctors diagnose Rett syndrome by observing signs and symptoms during the child's early growth and development and conducting ongoing evaluations of the child's physical and neurological status. Scientists have developed a genetic test to complement the clinical diagnosis, which involves searching for the MECP2 mutation on the child's X chromosome. A pediatric neurologist, clinical geneticist, or developmental pediatrician should be consulted to confirm the diagnosis of Rett syndrome.
Treating Rett syndrome
There is no cure for Rett syndrome. Treatment for the disorder is symptomatic—focusing on the management of symptoms—and supportive, requiring a multidisciplinary approach. Medication may be needed for breathing irregularities and motor difficulties, and anticonvulsant drugs may be used to control seizures. There should be regular monitoring for scoliosis and possible heart abnormalities. Occupational therapy can help children develop skills needed for performing self-directed activities (e.g., dressing, feeding, and practicing arts and crafts), while physical therapy and hydrotherapy may prolong mobility.
Treatment for the disorder is symptomatic—focusing on the management of symptoms—and supportive, requiring a multidisciplinary approach. Medication may be needed for breathing irregularities and motor difficulties, and anticonvulsant drugs may be used to control seizures. There should be regular monitoring for scoliosis and possible heart abnormalities. Occupational therapy can help children develop skills needed for performing self-directed activities (e.g., dressing, feeding, and practicing arts and crafts), while physical therapy and hydrotherapy may prolong mobility.
Some children may require special equipment and aids such as braces to arrest scoliosis, splints to modify hand movements, and nutritional programs to help them maintain adequate weight. Special academic, social, vocational, and support services may be required in some cases.
Despite the difficulties with symptoms, many individuals with Rett syndrome continue to live into middle age and beyond. Because the disorder is rare, very little is known about long-term prognosis and life expectancy.
What are the latest updates on Rett syndrome?
The National Institute of Neurological Disorders and Stroke (NINDS), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Institute of Mental Health (NIMH), and the Office of Rare Diseases Research (ORDR) support research on Rett syndrome.
Understanding the cause is necessary for developing new therapies to manage specific symptoms, as well as for providing better methods of diagnosis. The discovery of the main Rett syndrome gene (MECP2) in 1999 provides a basis for further genetic studies and enables the use of recently developed animal models such as transgenic mice which are deficient in MECP2. These mice have neurologic abnormalities that can be reversed by activating the MECP2 gene later in life.
One NINDS-supported study looks for mutations in the MECP2 gene of individuals with Rett syndrome to learn about MeCP2 protein function and dysfunction. Information from this study will increase understanding of the disorder and may lead to new therapies. Other research aims at identifying molecular pathways that are affected by the dysfunction, developing animal models of the disorder, and early-stage therapy development.
Other research aims at identifying molecular pathways that are affected by the dysfunction, developing animal models of the disorder, and early-stage therapy development.
Some researchers suggest that the specific type of mutation in the MECP2 gene affects the severity of symptoms of Rett syndrome. Studies are now underway to understand each mutation that may cause the features of Rett syndrome, and how these mutations might change the features of the syndrome. One NIH-funded study of the natural history of Rett syndrome should also provide new information about these topics.
Scientists know that lack of a properly functioning MeCP2 protein disturbs the function of mature brain cells, but they do not know the exact mechanisms by which this happens. Investigators are trying to find other genetic switches that operate in a similar way to the MeCP2 protein. Once they discover how the protein works and locate similar switches, they may devise therapies that can substitute for the malfunctioning switch. Another outcome might involve manipulating other biochemical pathways to compensate for the malfunctioning MECP2 gene, thereby preventing progression of the disorder. Gene therapy to achieve regulated expression of a normal MECP2 gene is also under study in animal models.
Another outcome might involve manipulating other biochemical pathways to compensate for the malfunctioning MECP2 gene, thereby preventing progression of the disorder. Gene therapy to achieve regulated expression of a normal MECP2 gene is also under study in animal models.
Researchers are also trying to find other genes that may be involved in Rett syndrome. Some studies have helped to narrow the search for these genes, but much is still unknown about how these genes may cause or contribute to Rett syndrome.
file-medical
Learn About Clinical Trials
Clinical trials are studies that allow us to learn more about disorders and improve care. They can help connect patients with new and upcoming treatment options.
How can I or my loved one help improve care for people with Rett syndrome?
Consider participating in a clinical trial so clinicians and scientists can learn more about Rett syndrome and related disorders. Clinical research uses human volunteers to help researchers learn more about a disorder and perhaps find better ways to safely detect, treat, or prevent disease.
All types of volunteers are needed—those who are healthy or may have an illness or disease—of all different ages, sexes, races, and ethnicities to ensure that study results apply to as many people as possible, and that treatments will be safe and effective for everyone who will use them.
For information about participating in clinical research visit NIH Clinical Research Trials and You. Learn about clinical trials currently looking for people with Rett syndrome at Clinicaltrials.gov.
Where can I find more information about Rett syndrome?
Information may be available from the following organizations:
Easter Seals
Phone: 312-726-6200 or 800-221-6827Genetic and Rare Diseases (GARD) Information Center
Phone: 888-205-2311International Rett Syndrome Foundation
Phone: 513-874-1298 or 800-818-7388National Institute of Child Health and Human Development (NICHD)
Phone: 301-496-5133National Institute of Mental Health (NIMH)
Phone: 301-443-4513, 866-415-8051 or 301-443-8431Rett Syndrome Research Trust
Phone: 203-445-0041
Rett syndrome.
 What is Rett Syndrome?
What is Rett Syndrome? IMPORTANT
The information in this section should not be used for self-diagnosis or self-treatment. In case of pain or other exacerbation of the disease, only the attending physician should prescribe diagnostic tests. For diagnosis and proper treatment, you should contact your doctor.
Rett syndrome is a genetic disease characterized by impaired development of the nervous system due to the lack of inhibition of certain genes. Manifestations of this condition are progressive mental retardation in girls (with extremely rare atypical forms - in boys), muscle hypotension, ataxia, spinal curvature. Diagnosis of Rett syndrome is based on data from a general and neurological examination, magnetic resonance imaging, electroencephalography, and molecular genetic analyzes. There is no specific treatment (there are only certain developments with encouraging results in animal experiments), symptomatic therapy is used to alleviate the patient's condition.
- Causes of Rett syndrome
- Rett syndrome symptoms
- Diagnostics
- Rett syndrome treatment
- Prognosis and prevention
- Prices for treatment
General
Rett syndrome is a genetic neuropsychiatric disease that almost always develops in girls and is manifested by a severe degree of mental retardation. This pathology was first identified back in 1954 by the Austrian neurologist A. Rett, however, he singled out this disease as a separate nosological unit only in 1966 year. Rett syndrome became widely known in the scientific world in 1983 after the studies of B. Hagberg. This condition is quite common - its occurrence is approximately 1:10-15 thousand newborn girls, in total, several tens of thousands of cases of pathology have been described to date. The mechanism of inheritance of Rett syndrome is dominant, linked to the X chromosome, which is why it occurs almost always in girls. In boys, due to the lack of a paired X chromosome, the genetic damage that leads to this disease is almost always fatal. However, there are several atypical forms of Rett syndrome, characterized by a smoother clinical picture and therefore affecting males. In addition, in boys, such a pathology can develop in the presence of an additional X chromosome - Klinefelter's syndrome.
However, there are several atypical forms of Rett syndrome, characterized by a smoother clinical picture and therefore affecting males. In addition, in boys, such a pathology can develop in the presence of an additional X chromosome - Klinefelter's syndrome.
Rett syndrome
Causes of Rett syndrome
The etiology and pathogenesis of Rett syndrome are quite complex and are due to the interaction of various genes and their influence on the development of the human brain. The root cause of the disease is a nonsense mutation (according to some reports, missense mutations also lead to similar disorders) of the MECP2 gene localized on the X chromosome, as a result of which its expression completely stops. It encodes a specific protein called methyl-CpG-binding protein 2, which is involved in the regulation of transcription of certain sections of DNA. This protein contains two domains, one of which facilitates its attachment to methylated regions of chromosomes (which are located near the genes that regulate brain development), and the second acts as a transcriptional repressor. The cause of Rett syndrome is precisely the lack of inhibition of certain genes, which leads to a violation of the formation of nervous tissue.
The cause of Rett syndrome is precisely the lack of inhibition of certain genes, which leads to a violation of the formation of nervous tissue.
At the same time, Rett syndrome cannot be considered as a neurodegenerative disease, since there is no destruction of neurons or neuroglia in it. Histological studies of brain tissues of patients reveal a violation of the ultrastructure of nerve cells - a decrease in size, a change in the number of dendrites, and difficult formation of nerve tissues. The volume of neuroglia in Rett syndrome is reduced, as a result, at the macroscopic level, the size of the brain also decreases by 20-30% compared to the age norm. One of the reasons for the above processes is the lack of inhibition of the release of the GAD67 enzyme (inhibition of the gene for this enzyme is carried out by methyl-CpG-binding protein 2), which, in turn, leads to an increase in the concentration of inhibitory transmitters from the GABA group. As a result, in patients with Rett syndrome, there is a significant prevalence of inhibitory processes in the brain, which affects not only the physiology of the central nervous system, but also its morphological structure.
Geneticists have found that the complete absence of the normal MECP2 gene in the genome in the vast majority of cases is a lethal condition and often leads to intrauterine death of the fetus. This condition occurs in boys (due to the presence of only one X chromosome) or in homozygous girls, which is extremely rare. Because of this, in the sexual distribution of Rett syndrome, there is an absolute prevalence of female patients. Mutations of the MECP2 gene in most cases are spontaneous or germline - presumably, 70% of cases of this disease are due to a genetic defect in the X chromosome in the father's germ cells. Defects in this gene also lead to other pathologies of the central nervous system - Zapel's variant, Loub's syndrome (X-linked mental retardation in boys), and congenital encephalopathy. Some researchers attribute these conditions to atypical forms of Rett syndrome.
Rett syndrome symptoms
In newborn girls, Rett syndrome at first does not manifest itself in any way; for the first 6-12 months, the development of the child occurs at a normal pace without any deviations. In the future, the progression of the disease is characterized by a certain staging. The first stage of Rett syndrome, most often occurring between the ages of 6 months and 2.5 years, is characterized by the appearance of muscle hypotonia in a child, a slowdown in psychomotor development, followed by lagging behind peers, loss of interest in games and people around. Pediatricians note a slower than normal growth of the feet and hands in length and a slowdown in the growth of head circumference. Sometimes, in addition to neurological manifestations, there may be a violation of the liver, heart, gastrointestinal tract.
In the future, the progression of the disease is characterized by a certain staging. The first stage of Rett syndrome, most often occurring between the ages of 6 months and 2.5 years, is characterized by the appearance of muscle hypotonia in a child, a slowdown in psychomotor development, followed by lagging behind peers, loss of interest in games and people around. Pediatricians note a slower than normal growth of the feet and hands in length and a slowdown in the growth of head circumference. Sometimes, in addition to neurological manifestations, there may be a violation of the liver, heart, gastrointestinal tract.
The second stage of Rett syndrome is characterized by more pronounced clinical manifestations. It develops within 1-2 years after the onset of the first symptoms of the disease, while the child first has anxiety, sleep disturbances. Then, quite quickly, in just a few weeks, patients with Rett syndrome lose almost all the skills acquired up to this time - speech is lost, the ability to walk disappears. Also, this stage of the development of the pathology is characterized by respiratory disorders - periods of apnea of 1-2 minutes can be interspersed with attacks of rapid and deep respiratory movements (hyperventilation). Respiratory disorders in Rett syndrome are distinguished by the presence only when the patient is awake and the absence during sleep. Often there are numerous neurological disorders: ataxia, epileptic seizures, often repetitive stereotyped movements.
Also, this stage of the development of the pathology is characterized by respiratory disorders - periods of apnea of 1-2 minutes can be interspersed with attacks of rapid and deep respiratory movements (hyperventilation). Respiratory disorders in Rett syndrome are distinguished by the presence only when the patient is awake and the absence during sleep. Often there are numerous neurological disorders: ataxia, epileptic seizures, often repetitive stereotyped movements.
The third stage of Rett syndrome is called pseudo-stationary, since it shows little signs of progression of the disease. Usually it lasts from 4 to 15 years, the condition of patients is stable, however, there are convulsive seizures, severe mental retardation, hyperkinesis. In most cases of Rett syndrome, this stage ends in puberty. The fourth stage of the Rett syndrome is characterized by a decrease in the frequency of epileptic seizures up to their disappearance with the progression of movement disorders. Most patients completely lose their mobility, there is muscle atrophy, vascular disorders in the lower extremities, which can lead to the development of trophic ulcers. Due to the weakness of the muscular corset of the back in Rett syndrome, scoliosis or other forms of curvature of the spine occur.
Most patients completely lose their mobility, there is muscle atrophy, vascular disorders in the lower extremities, which can lead to the development of trophic ulcers. Due to the weakness of the muscular corset of the back in Rett syndrome, scoliosis or other forms of curvature of the spine occur.
Diagnostics
Diagnosis of Rett syndrome is made on the basis of a study of the patient's history, his current status, magnetic resonance imaging and encephalography, and molecular genetic analyzes. The study of hereditary history, as a rule, does not make much sense due to the sporadic nature of MECP2 gene mutations. Characteristic of the Rett syndrome are the normal development of the child up to 6-12 months, the occurrence of muscle hypotonia and anxiety in early childhood, the appearance of ataxia and frequent epileptic seizures in the future, and the rapid loss of acquired skills. In the future, patients recorded severe mental retardation, muscle weakness (up to atrophy), curvature of the spine, convulsive seizures.
When examining patients with Rett syndrome, growth retardation and its stop, a sharp decrease in head circumference, and the absence of speech are revealed (echolalia is characteristic at the initial stages of the pathology). Magnetic resonance imaging of the brain reveals a decrease in the size of the organ, fuzzy differentiation of gray and white matter, basal ganglia, and a decrease in the folding of the cerebral cortex. An electroencephalogram confirms a decrease in the background activity of the brain and a sharply weakened response to external stimuli.
The most accurate diagnostic information is provided by methods of modern genetics - the search for deletions in the MECP2 gene locus or direct sequencing of its sequence to determine mutations. Such confirmation of Rett syndrome is also possible within the framework of prenatal diagnosis of genetic diseases. An auxiliary role in establishing this condition can be played by an examination of internal organs (for example, by ultrasound methods) - in 20-30% of patients, underdevelopment of the liver or spleen is detected.
Rett syndrome treatment
There is currently no specific treatment for Rett syndrome. There are encouraging data from some research laboratories, whose employees were able to "turn on" the MECP2 gene in mice and thereby achieve the disappearance of the symptoms of the disease. In the field of practical medicine, only symptomatic therapy is still available, however, it is also associated with a number of difficulties - in particular, epileptic seizures in this disease are difficult to eliminate with anticonvulsants. Also, nootropic drugs are used to treat Rett syndrome, sleep disorders are corrected with sleeping pills from the barbiturate group or melatonin.
Prognosis and prevention
The prognosis of Rett syndrome is unfavorable, as this disease steadily leads to severe mental retardation, as well as to a number of motor and neurological disorders. Patients with this pathology, with appropriate care and symptomatic treatment, are able to live up to 40-50 years, but their risk of sudden death is quite high. The presence of malformations of internal organs significantly worsens the prognosis of Rett syndrome and reduces the life expectancy of patients, which occurs in about a third of cases. The main cause of death is respiratory or multiple organ failure and sudden death, in adult patients the risk of stroke is also high. Prevention of Rett syndrome is possible only in the form of prenatal diagnosis of this disease by genetic methods. In the presence of a defective form of the MECP2 gene in a boy, a violation of the formation of the brain and internal organs can be seen during preventive ultrasound examinations during pregnancy.
The presence of malformations of internal organs significantly worsens the prognosis of Rett syndrome and reduces the life expectancy of patients, which occurs in about a third of cases. The main cause of death is respiratory or multiple organ failure and sudden death, in adult patients the risk of stroke is also high. Prevention of Rett syndrome is possible only in the form of prenatal diagnosis of this disease by genetic methods. In the presence of a defective form of the MECP2 gene in a boy, a violation of the formation of the brain and internal organs can be seen during preventive ultrasound examinations during pregnancy.
You can share your medical history, what helped you in the treatment of Rett syndrome.
Sources
- In case of pain or other exacerbation of the disease, only the attending physician should prescribe diagnostic tests. For diagnosis and proper treatment, you should contact your doctor.
Rett Syndrome (SR)
In 1966 Austrian pediatrician Andreas Rett described 2 girls with a similar condition.
 In 1983, Bengt Hagberg and Jean Aicardi published the first paper on 35 cases in English. In 1984, the formation of the IRSA, and in 1985 the first congress of doctors on the problem of Rett syndrome was held.
In 1983, Bengt Hagberg and Jean Aicardi published the first paper on 35 cases in English. In 1984, the formation of the IRSA, and in 1985 the first congress of doctors on the problem of Rett syndrome was held. Rett Syndrome is a progressive hereditary disease that manifests itself in early childhood and is characterized by regression of psychomotor development, autistic features, loss of motor skills and specific motor automatisms, and with the frequent development of epilepsy.
Genetics SR:
- SR is characterized by impaired brain development caused by a mutation of the MECP2 transcription regulator gene encoding Methyl-CpG binding protein-2 on the X chromosome, the Xq28 locus (Schaner et al., 2004 ). 8 major mutations of this gene are known, which are detected in 80% of patients with SR
- The MECP2 gene regulates the processes of maturation of the central nervous system. The mutation leads to regression of psychomotor development and mental disorders, in particular autistic behavior (Huppke & Cartner, 2005)
- Most reported cases worldwide are sporadic, less than 1% are familial
SR prevalence:
- 1-3.
 5 cases per 10,000 female population
5 cases per 10,000 female population - 10% of cases among mentally retarded female patients
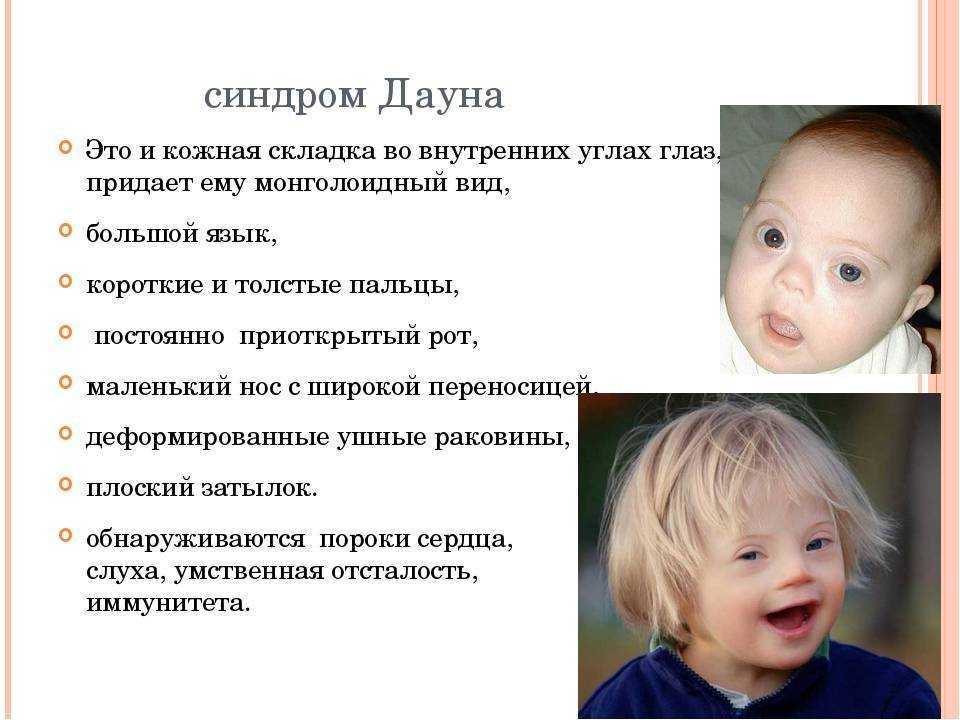
- About 30% of cases of progressive dementia with early onset in girls, the second most common (after Down's syndrome) hereditary disease with dementia occurring in girls (Aicardi, 1998)
Debut and course of SR:
- Debut of the disease in the interval from 6 to 20 months.
- Stage 1 (stagnation) from 6 months. up to 1.5 years. Psychomotor development stops, head growth slows down
- stage 2 (rapid regression), 1.5 to 3 years. Regression with loss of purposeful manual praxis and appearance of manual stereotypes: “washing”, “twisting”, “rubbing”, “hands to mouth”
- Stage 3 (pseudo-stationary). Stabilization of severe mental retardation with autistic behavior
- stage 4 (late movement disorders). After 8 years. Stable severe intellectual disorders. Increasing movement disorders
Rett syndrome criteria (J Aicardi 1998, Percy & Lane 2005)
- Normal perinatal period
- Normal head circumference measurements at birth
- Relatively normal psychomotor development up to 6 months.

- Postnatal head growth retardation from 5 months. up to 4 years
- Loss of purposeful manual activity occurring between 6 and 30 months, associated with impaired communication and loss of social skills
- Loss of speech associated with severe mental retardation
- Appearance of stereotyped manual movements. These manifestations occur following the loss of purposeful hand movements
- Stereotyped movements in the facial muscles: chewing, tapping teeth, baring teeth
- The appearance of trunk ataxia and apraxia of walking at 1-4 years
Epilepsy in SR:
Paroxysmal disorders in SR: hyperventilatory attacks, vaso-vagal syncope and epilepsy (J Aicardi 1998). The debut of epilepsy in SR at the age of 5-15 years.
Characteristics of epileptic seizures in SR:
Type 1 - early form (debut from 10 days to 10 months) with frequent generalized seizures and infantile spasms.
 In the future, seizures have polymorphism and resistance to therapy.
In the future, seizures have polymorphism and resistance to therapy. type 2 - late childhood form (debut 1.5-6 years). Rare tonic-clonic seizures and myoclonic, atonic, atypical absences predominate.
Reflex attacks in SR: food provocation, stress, hyperventilation. Often - autoinduction.
Features of the course and prognosis of epilepsy depend on the nature of the mutation in SR:
- Early onset has a severe course of epilepsy.
- Late childhood debut - rare seizures and a high percentage of remission.
- In some favorable cases, after 8-10 years, there is stabilization and a decrease in the frequency of attacks, even without therapy.
Features of the EEG in SR:
Regional and multiregional activity of an acute-slow wave, diffuse peak-wave discharges of a low degree of synchronization, the morphology of the complexes can be of the BEPD type.
Treatment of epilepsy in SR:
- At the onset of infantile spasms, Sabril (Vigabatrin) at an average dose of 100 mg/kg/day
- In atypical absences, myoclonic and atonic seizures - valproates (depakin chrono, depakin chronosphere, convulex retard) in monotherapy 30-60 mg/kg/day.